

Functional genomic landscape of cancer-intrinsic evasion of killing by T cells
- PMID: 32968282
- PMCID: PMC9014559
- DOI: 10.1038/s41586-020-2746-2
Functional genomic landscape of cancer-intrinsic evasion of killing by T cells
Abstract
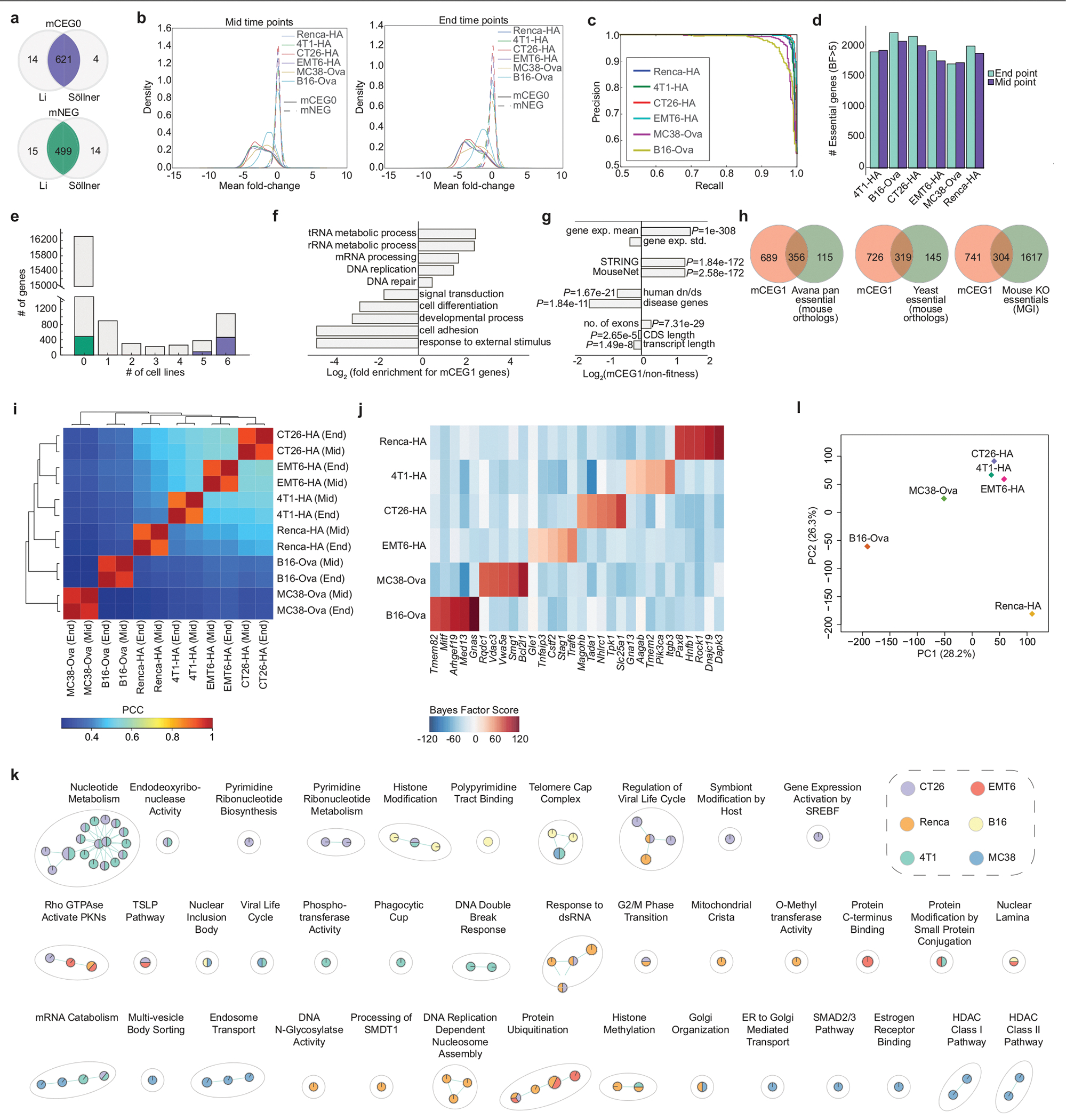
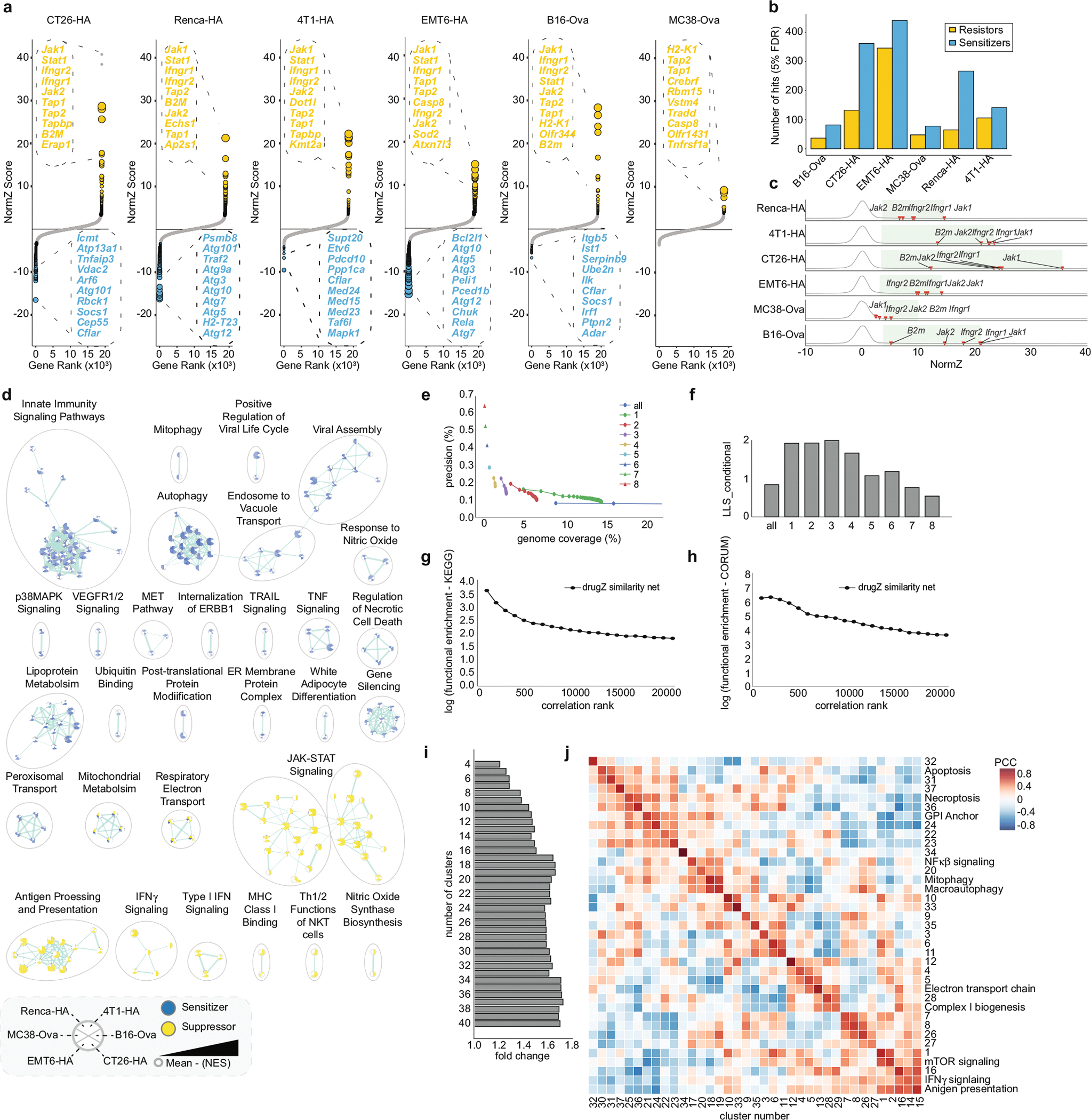
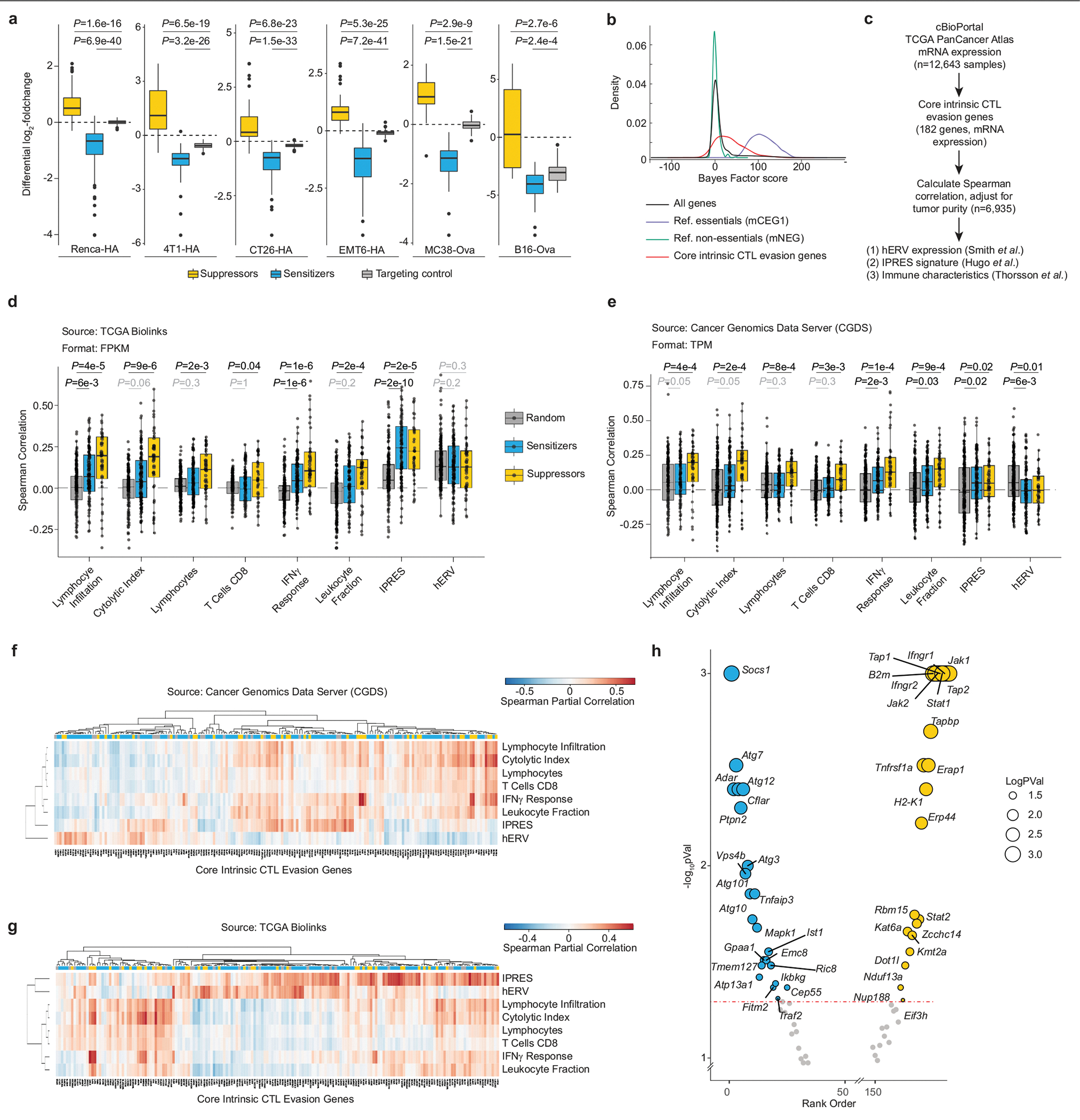
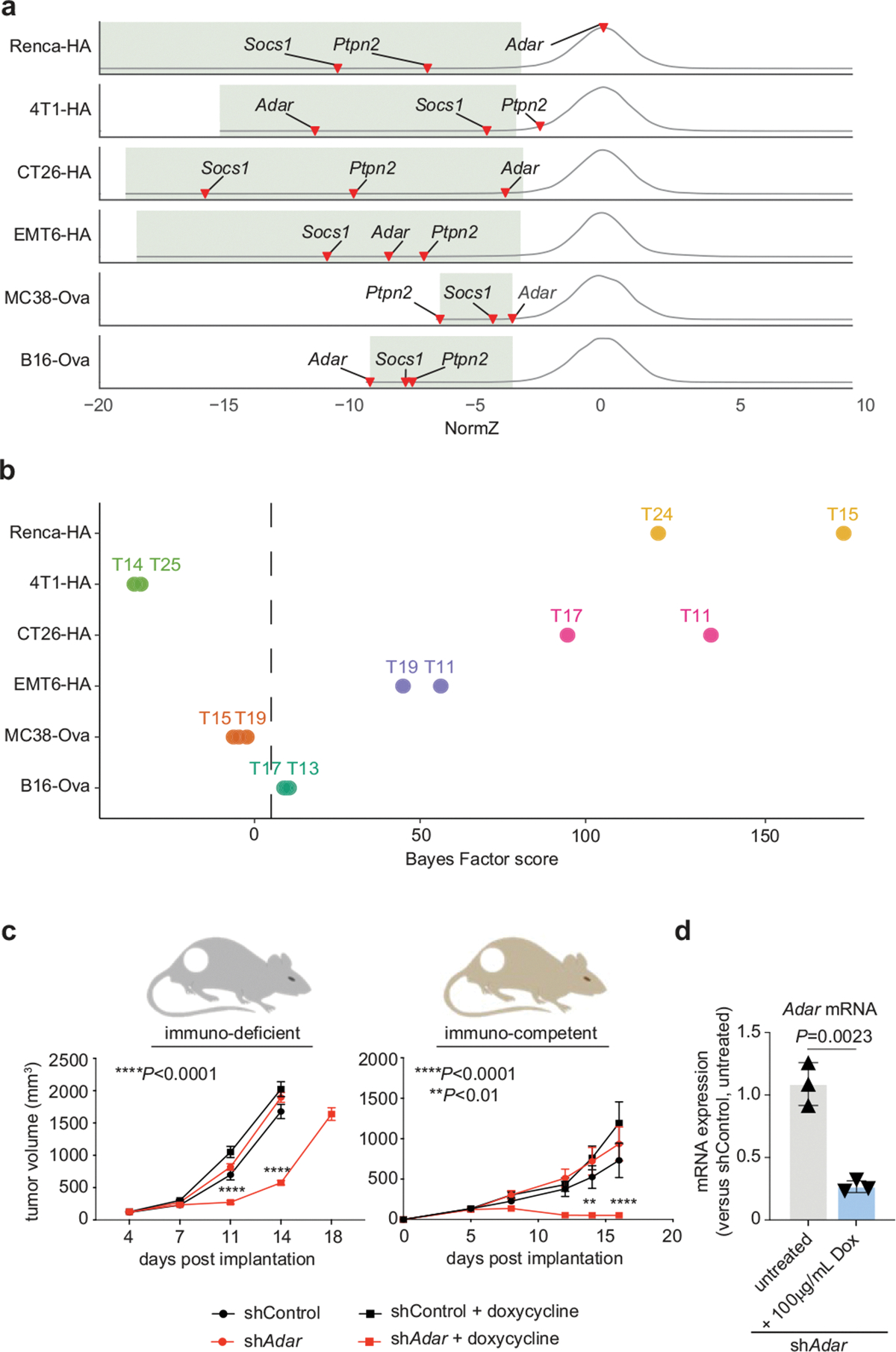
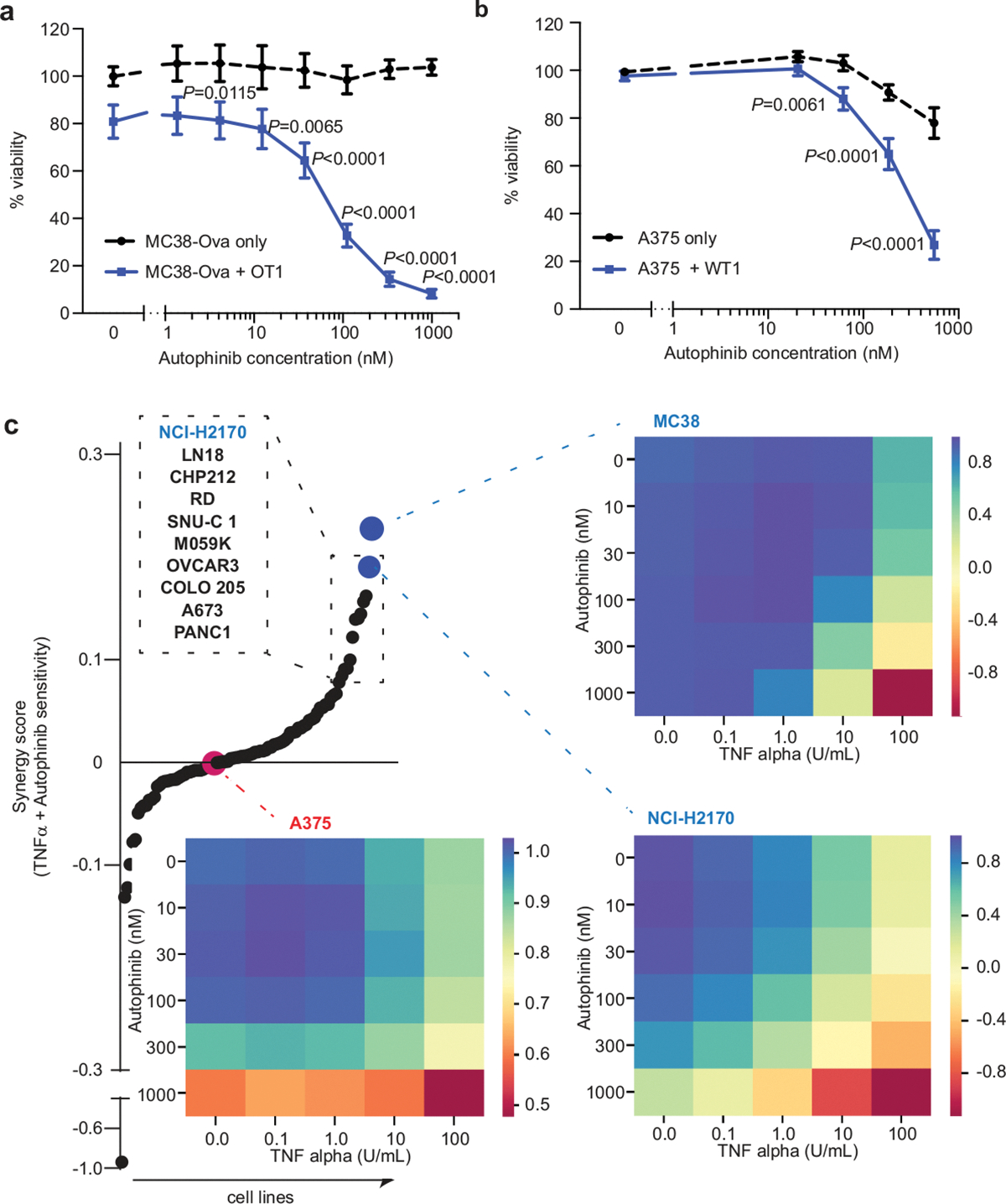
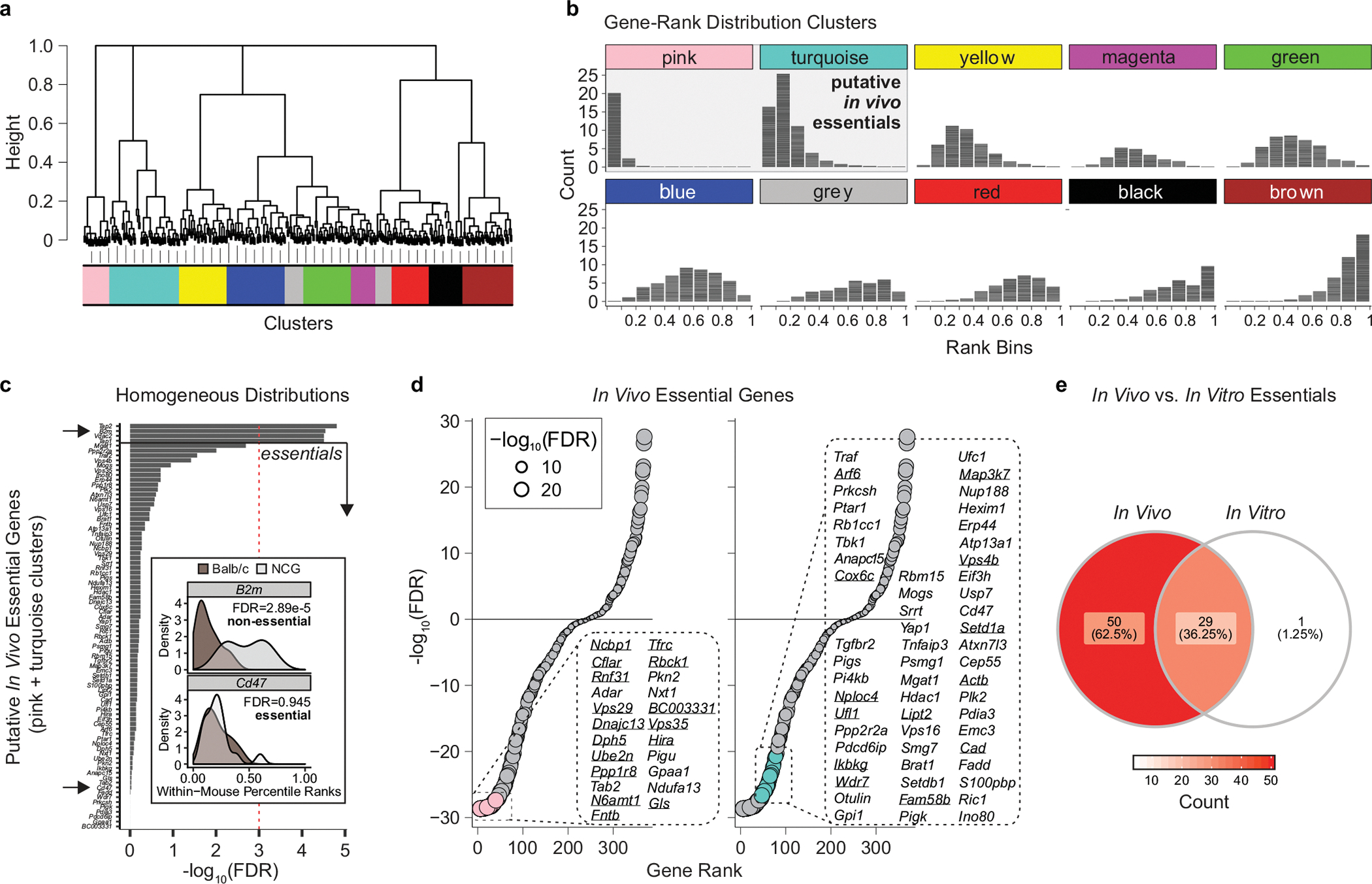
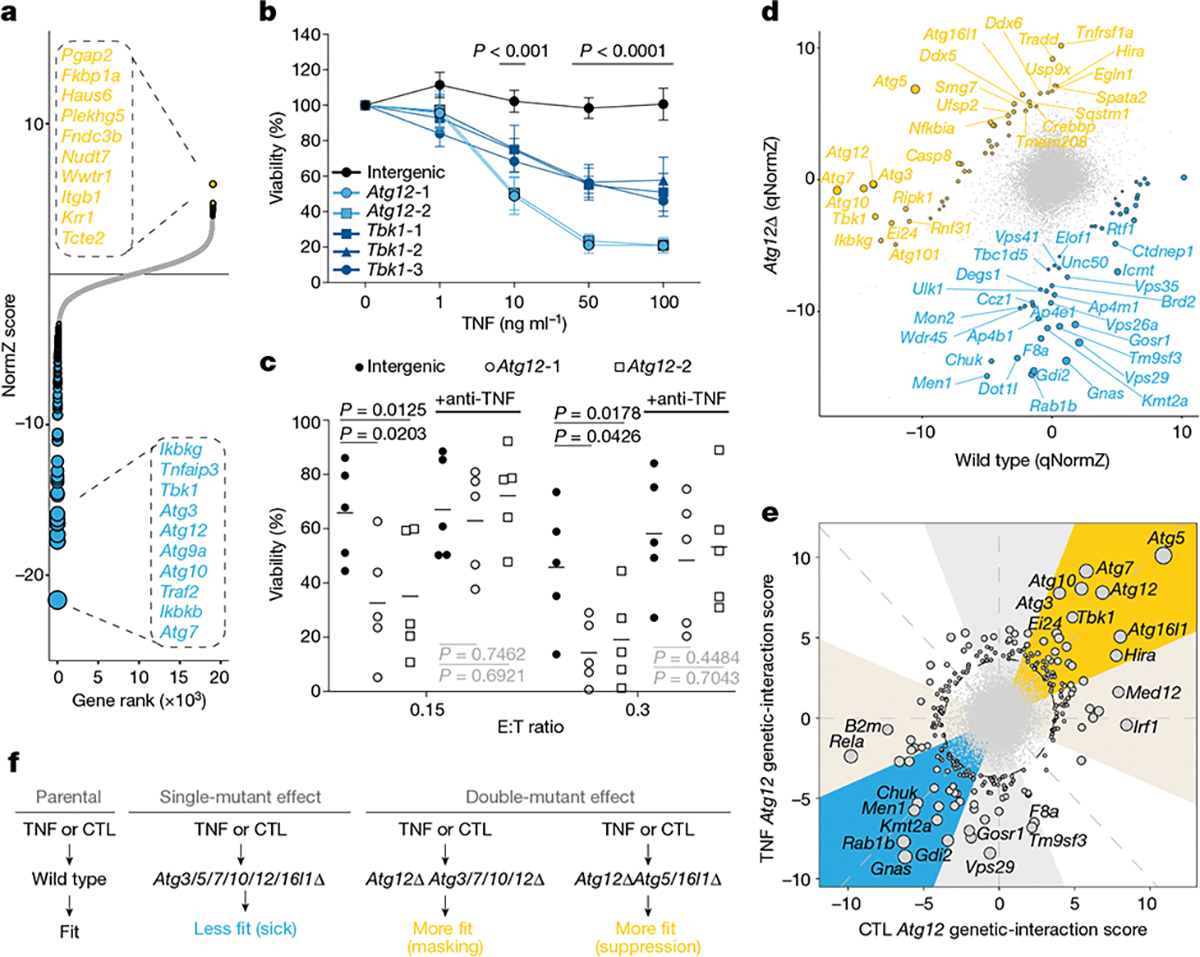
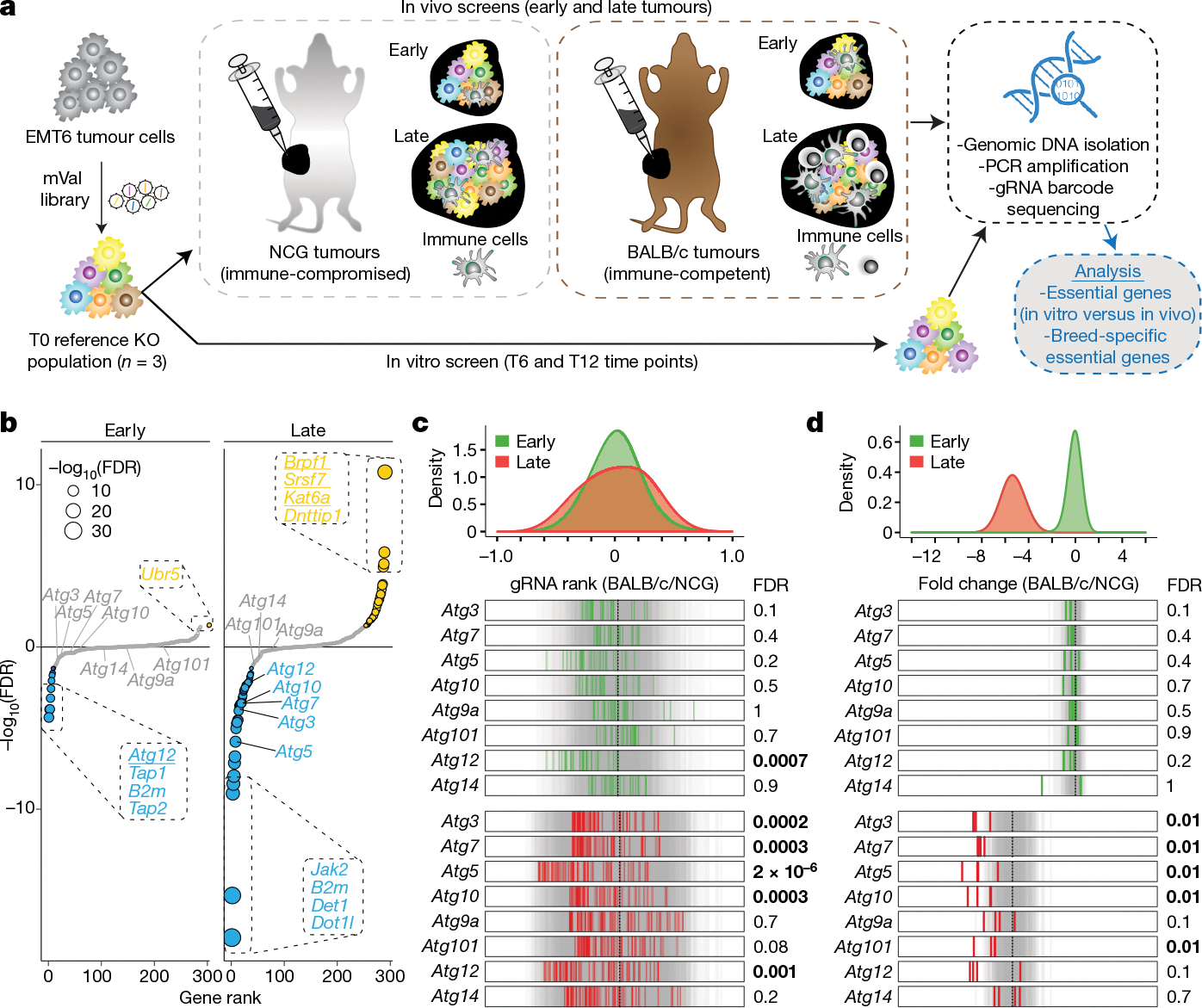
The genetic circuits that allow cancer cells to evade destruction by the host immune system remain poorly understood1-3. Here, to identify a phenotypically robust core set of genes and pathways that enable cancer cells to evade killing mediated by cytotoxic T lymphocytes (CTLs), we performed genome-wide CRISPR screens across a panel of genetically diverse mouse cancer cell lines that were cultured in the presence of CTLs. We identify a core set of 182 genes across these mouse cancer models, the individual perturbation of which increases either the sensitivity or the resistance of cancer cells to CTL-mediated toxicity. Systematic exploration of our dataset using genetic co-similarity reveals the hierarchical and coordinated manner in which genes and pathways act in cancer cells to orchestrate their evasion of CTLs, and shows that discrete functional modules that control the interferon response and tumour necrosis factor (TNF)-induced cytotoxicity are dominant sub-phenotypes. Our data establish a central role for genes that were previously identified as negative regulators of the type-II interferon response (for example, Ptpn2, Socs1 and Adar1) in mediating CTL evasion, and show that the lipid-droplet-related gene Fitm2 is required for maintaining cell fitness after exposure to interferon-γ (IFNγ). In addition, we identify the autophagy pathway as a conserved mediator of the evasion of CTLs by cancer cells, and show that this pathway is required to resist cytotoxicity induced by the cytokines IFNγ and TNF. Through the mapping of cytokine- and CTL-based genetic interactions, together with in vivo CRISPR screens, we show how the pleiotropic effects of autophagy control cancer-cell-intrinsic evasion of killing by CTLs and we highlight the importance of these effects within the tumour microenvironment. Collectively, these data expand our knowledge of the genetic circuits that are involved in the evasion of the immune system by cancer cells, and highlight genetic interactions that contribute to phenotypes associated with escape from killing by CTLs.
Conflict of interest statement
Figures






Comment in
-
Inhibition of tumor autophagy: a strategy to improve anti-tumor immunity?Signal Transduct Target Ther. 2020 Dec 4;5(1):286. doi: 10.1038/s41392-020-00429-8. Signal Transduct Target Ther. 2020. PMID: 33277468 Free PMC article. No abstract available.
Similar articles
-
Complementary CRISPR screen highlights the contrasting role of membrane-bound and soluble ICAM-1 in regulating antigen-specific tumor cell killing by cytotoxic T cells.Elife. 2023 Sep 21;12:e84314. doi: 10.7554/eLife.84314. Elife. 2023. PMID: 37732732 Free PMC article.
-
Tumor immune evasion arises through loss of TNF sensitivity.Sci Immunol. 2018 May 18;3(23):eaar3451. doi: 10.1126/sciimmunol.aar3451. Sci Immunol. 2018. PMID: 29776993
-
Molecular mechanism of MART-1+/A*0201+ human melanoma resistance to specific CTL-killing despite functional tumor-CTL interaction.Cancer Res. 2011 Feb 15;71(4):1406-17. doi: 10.1158/0008-5472.CAN-10-1296. Epub 2010 Dec 15. Cancer Res. 2011. Retraction in: Cancer Res. 2017 Jul 1;77(13):3718. doi: 10.1158/0008-5472.CAN-17-0556 PMID: 21159666 Free PMC article. Retracted.
-
CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review.J Cell Physiol. 2019 Jun;234(6):8509-8521. doi: 10.1002/jcp.27782. Epub 2018 Nov 22. J Cell Physiol. 2019. PMID: 30520029 Review.
-
Natural selection of tumor variants in the generation of "tumor escape" phenotypes.Nat Immunol. 2002 Nov;3(11):999-1005. doi: 10.1038/ni1102-999. Nat Immunol. 2002. PMID: 12407407 Free PMC article. Review.
Cited by
-
Molecular profiling of core immune-escape genes highlights LCK as an immune-related prognostic biomarker in melanoma.Front Immunol. 2022 Oct 20;13:1024931. doi: 10.3389/fimmu.2022.1024931. eCollection 2022. Front Immunol. 2022. PMID: 36341345 Free PMC article.
-
DNA barcoding reveals ongoing immunoediting of clonal cancer populations during metastatic progression and immunotherapy response.Nat Commun. 2022 Nov 7;13(1):6539. doi: 10.1038/s41467-022-34041-x. Nat Commun. 2022. PMID: 36344500 Free PMC article.
-
Arrest and Attack: Microtubule-Targeting Agents and Oncolytic Viruses Employ Complementary Mechanisms to Enhance Anti-Tumor Therapy Efficacy.Genes (Basel). 2024 Sep 11;15(9):1193. doi: 10.3390/genes15091193. Genes (Basel). 2024. PMID: 39336785 Free PMC article. Review.
-
Construction of an Immune Escape-Related Signature in Clear Cell Renal Cell Carcinoma and Identification of the Relationship between IFNAR1 and Immune Infiltration by Multiple Immunohistochemistry.Cancers (Basel). 2022 Dec 28;15(1):169. doi: 10.3390/cancers15010169. Cancers (Basel). 2022. PMID: 36612165 Free PMC article.
-
Caspase-8 contributes to an immuno-hot microenvironment by promoting phagocytosis via an ecto-calreticulin-dependent mechanism.Exp Hematol Oncol. 2023 Jan 12;12(1):7. doi: 10.1186/s40164-022-00371-1. Exp Hematol Oncol. 2023. PMID: 36635765 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
