

Procyanidin B2 mitigates endothelial endoplasmic reticulum stress through a PPARδ-Dependent mechanism
- PMID: 32961442
- PMCID: PMC7509074
- DOI: 10.1016/j.redox.2020.101728
Procyanidin B2 mitigates endothelial endoplasmic reticulum stress through a PPARδ-Dependent mechanism
Abstract
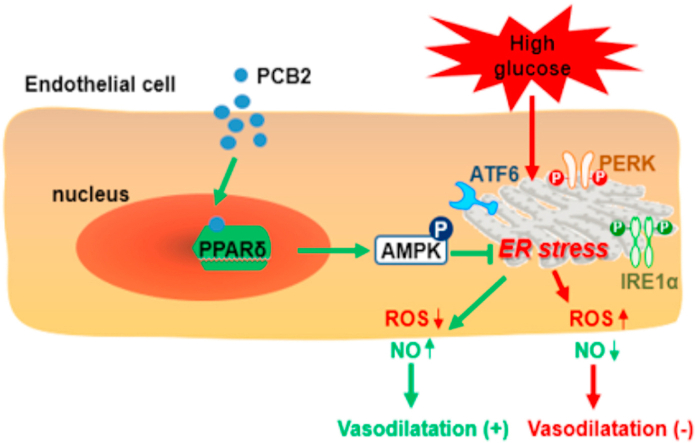
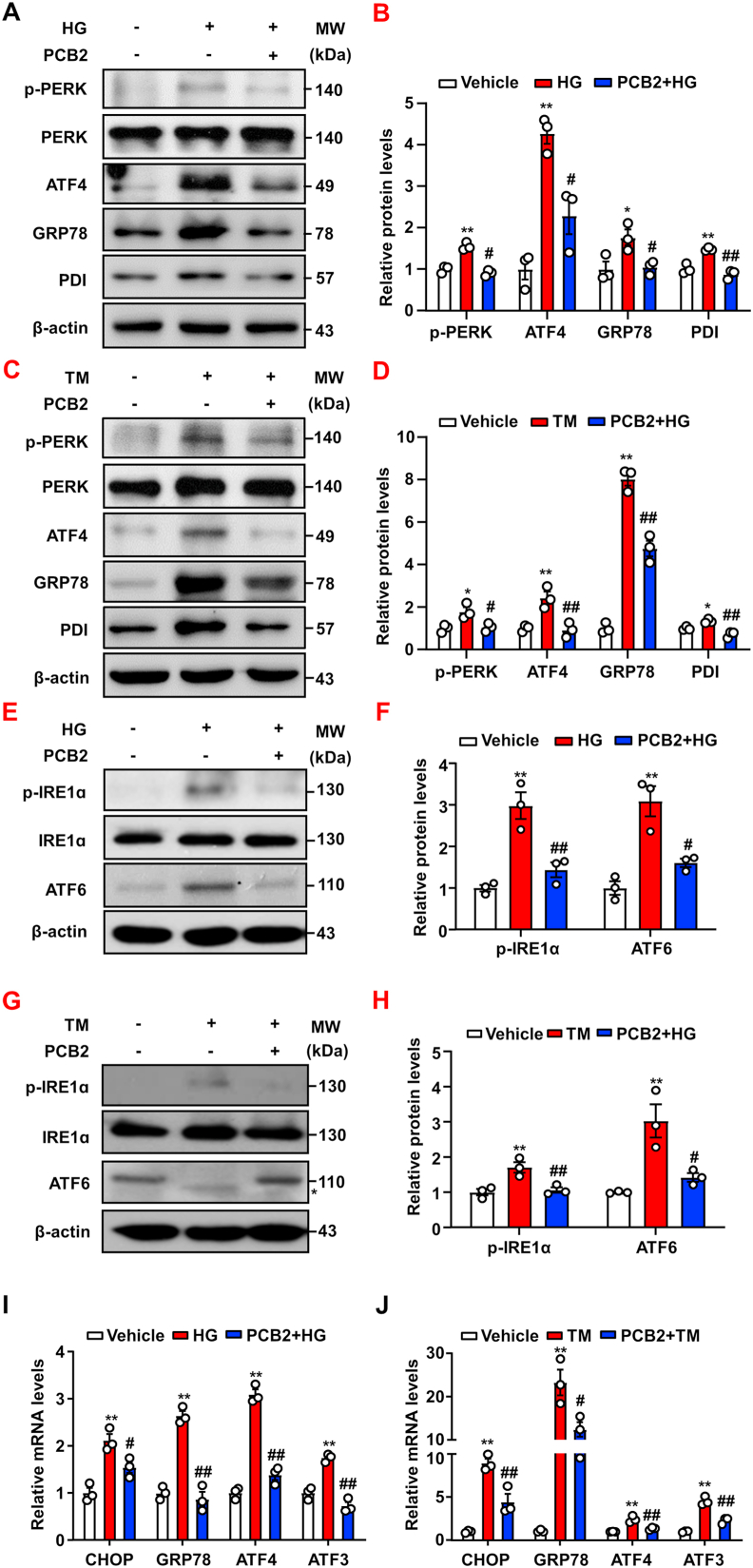
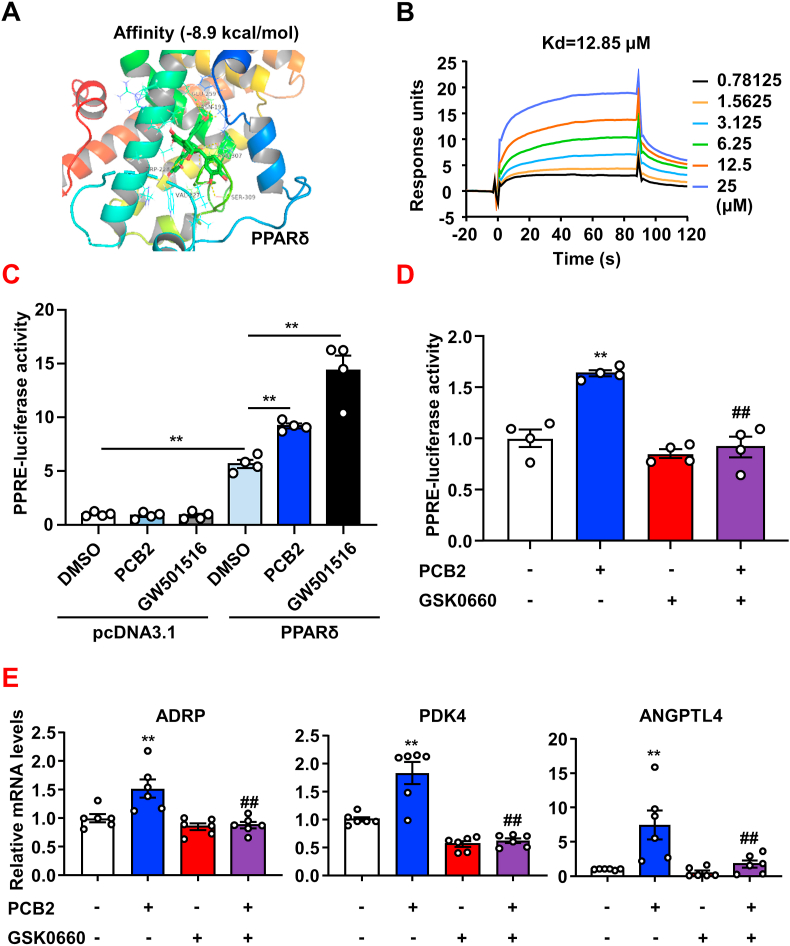
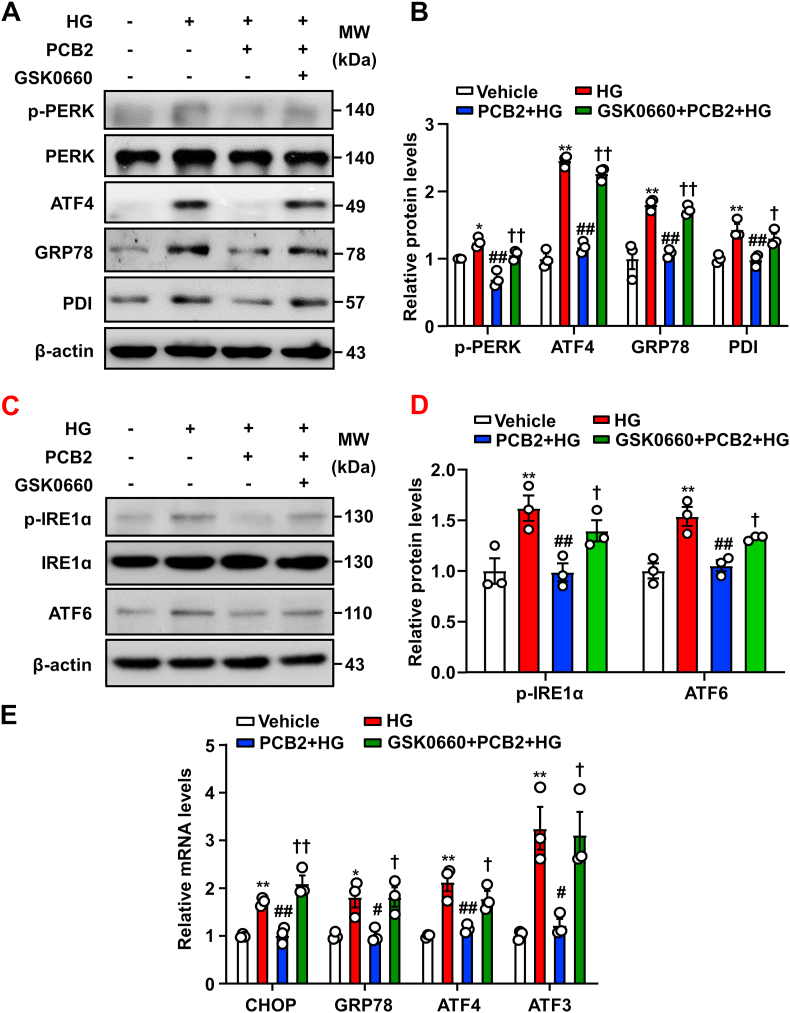
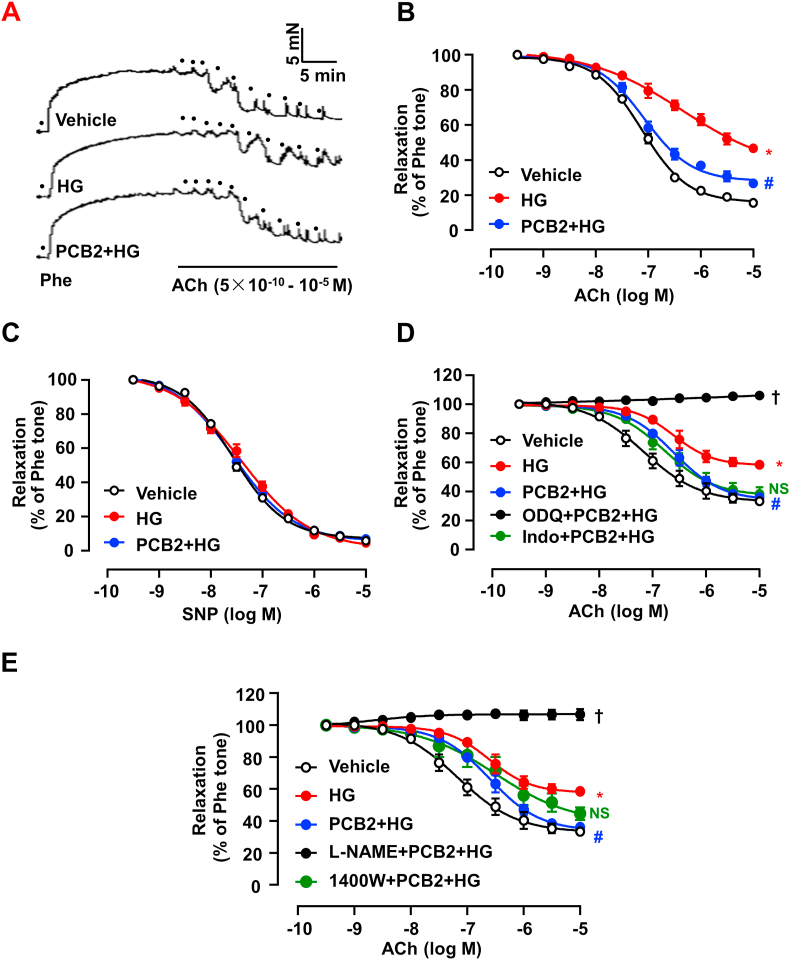
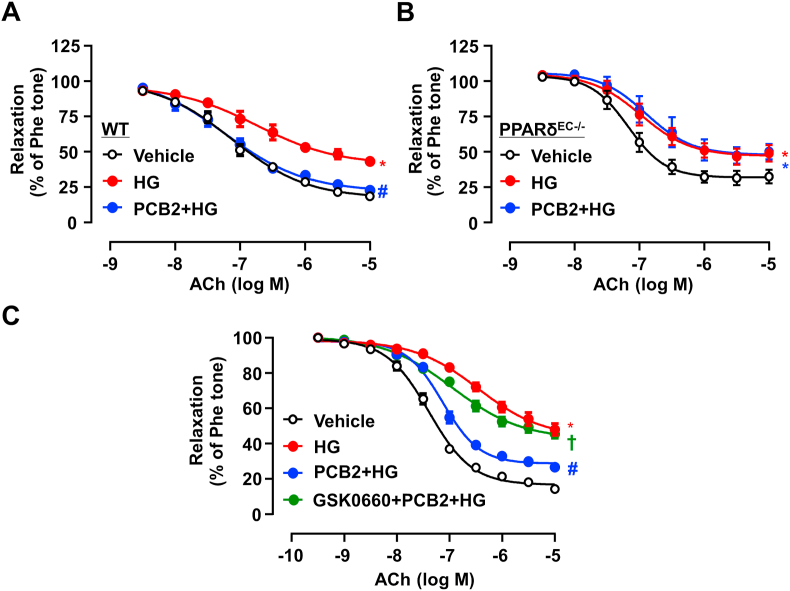
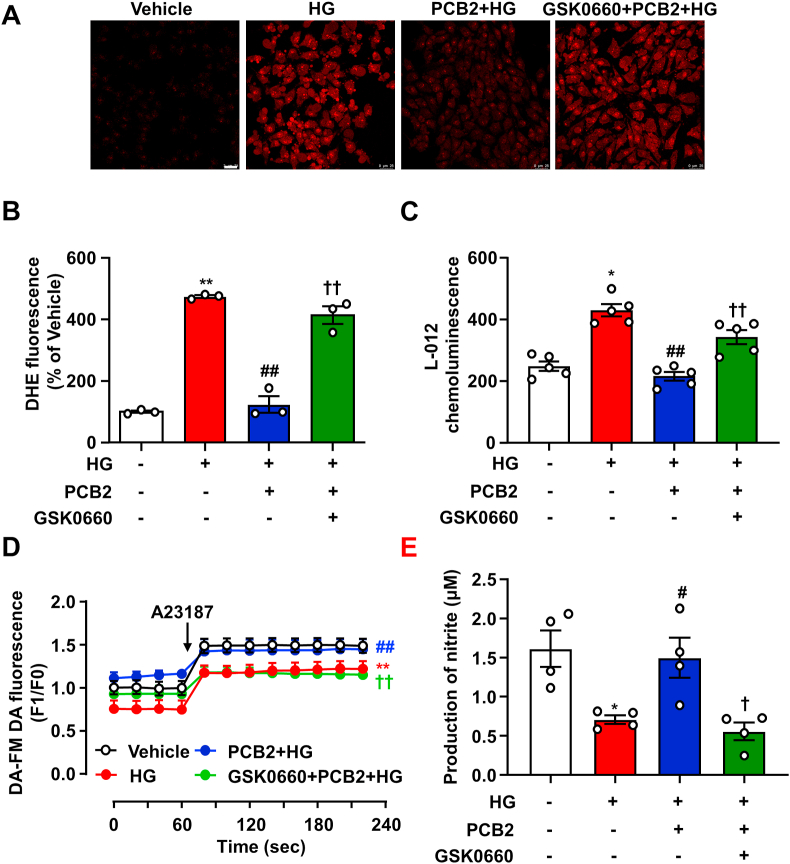
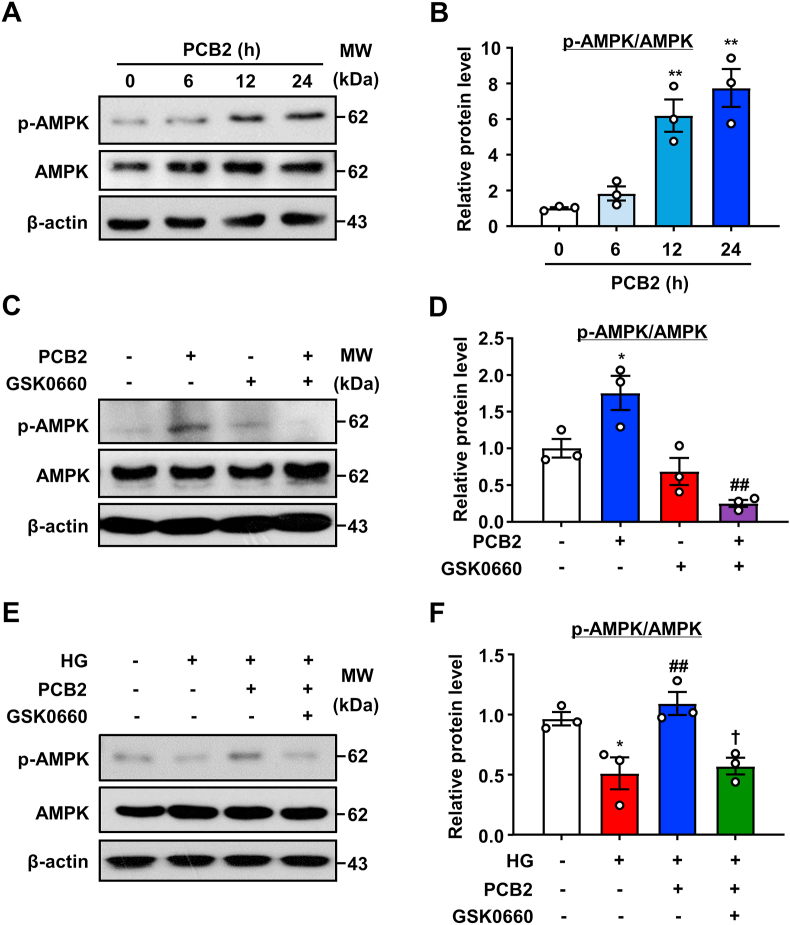
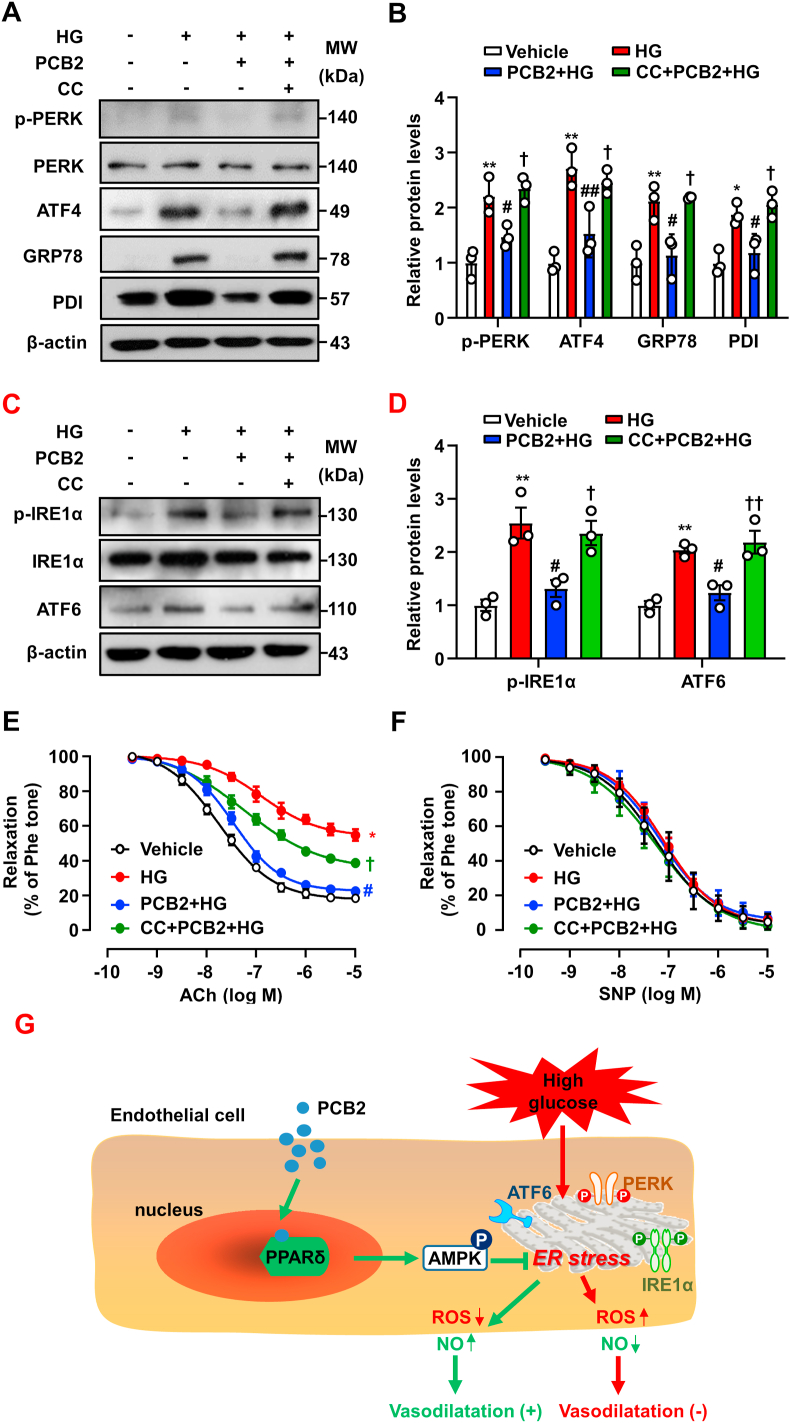
Hyperglycemia-induced endothelial endoplasmic reticulum (ER) stress is implicated in the pathophysiology of diabetes and its vascular complications. Procyanidins are enriched in many plant foods and have been demonstrated to exert several beneficial effects on diabetes, cardiovascular and other metabolic diseases. In the present study, we investigated the effect of procyanidin B2 (PCB2), the most widely distributed natural procyanidin, on ER stress evoked by high glucose in endothelial cells (ECs) and the underlying mechanisms. We showed that PCB2 mitigated the high glucose-activated ER stress pathways (PERK, IRE1α and ATF6) in human vascular ECs. In addition, we found that PCB2 attenuated endothelial ER stress via the activation of peroxisome proliferator-activated receptor δ (PPARδ). We demonstrated that PCB2 directly bound to and activated PPARδ. Conversely, GSK0660, a selective PPARδ antagonist, attenuated the suppressive effect of PCB2 on the ER stress signal pathway. Functionally, PCB2 ameliorated the high glucose-impaired endothelium-dependent relaxation in mouse aortas. The protective effect of PCB2 on vasodilation was abolished in the aortas pretreated with GSK0660 or those from the EC-specific PPARδ knockout mice. Moreover, the protective effects of PCB2 on ER stress and endothelial dysfunction required the inter-dependent actions of PPARδ and AMPK. Collectively, we demonstrated that PCB2 mitigated ER stress and ameliorated vasodilation via a PPARδ-mediated mechanism beyond its classic action as a scavenger of free radicals. These findings further highlighted the novel roles of procyanidins in intervening the ER stress and metabolic disorders related to endothelial dysfunction.
Keywords: Endoplasmic reticulum stress; Endothelium-dependent relaxation; Peroxisome proliferator-activated receptor δ; Procyanidin B2.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Paeonol protects against endoplasmic reticulum stress-induced endothelial dysfunction via AMPK/PPARδ signaling pathway.Biochem Pharmacol. 2016 Sep 15;116:51-62. doi: 10.1016/j.bcp.2016.07.013. Epub 2016 Jul 20. Biochem Pharmacol. 2016. PMID: 27449753
-
Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5' adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway.Arterioscler Thromb Vasc Biol. 2014 Apr;34(4):830-6. doi: 10.1161/ATVBAHA.113.301938. Epub 2014 Jan 30. Arterioscler Thromb Vasc Biol. 2014. PMID: 24482374
-
Omentin-1 protects against high glucose-induced endothelial dysfunction via the AMPK/PPARδ signaling pathway.Biochem Pharmacol. 2020 Apr;174:113830. doi: 10.1016/j.bcp.2020.113830. Epub 2020 Jan 28. Biochem Pharmacol. 2020. PMID: 32001235
-
Procyanidin B2 inhibits NLRP3 inflammasome activation in human vascular endothelial cells.Biochem Pharmacol. 2014 Dec 15;92(4):599-606. doi: 10.1016/j.bcp.2014.10.001. Epub 2014 Oct 23. Biochem Pharmacol. 2014. PMID: 25450671
-
Procyanidin B2 Activates PPARγ to Induce M2 Polarization in Mouse Macrophages.Front Immunol. 2019 Aug 7;10:1895. doi: 10.3389/fimmu.2019.01895. eCollection 2019. Front Immunol. 2019. PMID: 31440258 Free PMC article.
Cited by
-
Mitochondrial PKM2 deacetylation by procyanidin B2-induced SIRT3 upregulation alleviates lung ischemia/reperfusion injury.Cell Death Dis. 2022 Jul 11;13(7):594. doi: 10.1038/s41419-022-05051-w. Cell Death Dis. 2022. PMID: 35821123 Free PMC article.
-
Paeonol protects against doxorubicin-induced cardiotoxicity by promoting Mfn2-mediated mitochondrial fusion through activating the PKCε-Stat3 pathway.J Adv Res. 2023 May;47:151-162. doi: 10.1016/j.jare.2022.07.002. Epub 2022 Jul 14. J Adv Res. 2023. PMID: 35842187 Free PMC article.
-
Bevacizumab-Induced Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, and ERK Inactivation Contribute to Cardiotoxicity.Oxid Med Cell Longev. 2021 Mar 17;2021:5548130. doi: 10.1155/2021/5548130. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 33859777 Free PMC article.
-
The Cell Protective Effect of Adenine on Hypoxia-Reoxygenation Injury through PPAR Delta Activation.Life (Basel). 2021 Dec 16;11(12):1408. doi: 10.3390/life11121408. Life (Basel). 2021. PMID: 34947939 Free PMC article.
-
Research progress of procyanidins in repairing cartilage injury after anterior cruciate ligament tear.Heliyon. 2024 Feb 18;10(4):e26070. doi: 10.1016/j.heliyon.2024.e26070. eCollection 2024 Feb 29. Heliyon. 2024. PMID: 38420419 Free PMC article. Review.
References
-
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. - PubMed
-
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. - PubMed
-
- Battson M.L., Lee D.M., Gentile C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017;312:H355–H367. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
