

Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40
- PMID: 32631408
- PMCID: PMC7339583
- DOI: 10.1186/s13195-020-00649-8
Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40
Abstract
Background: Cathepsin D (CatD) is a lysosomal protease that degrades both the amyloid β-protein (Aβ) and the microtubule-associated protein, tau, and has been genetically linked to late-onset Alzheimer disease (AD). Here, we sought to examine the consequences of genetic deletion of CatD on Aβ proteostasis in vivo and to more completely characterize the degradation of Aβ42 and Aβ40 by CatD.
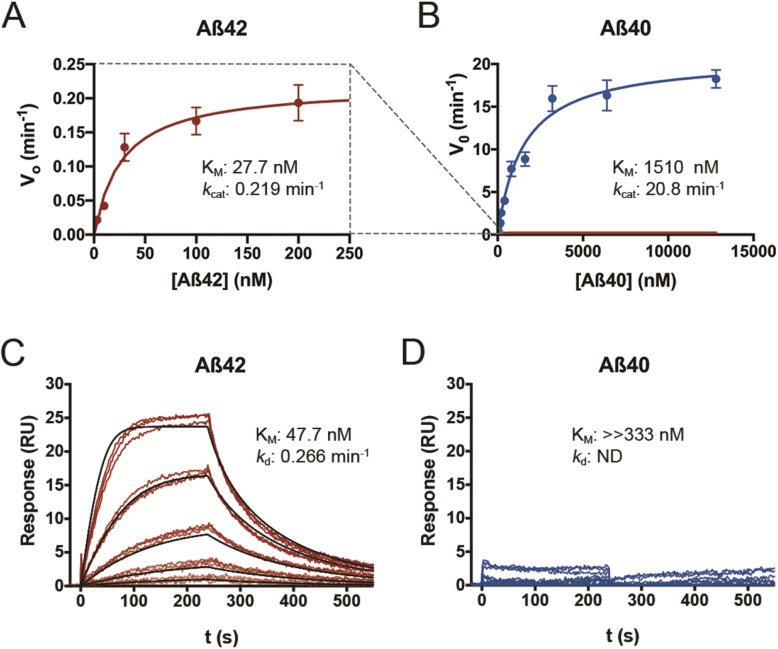
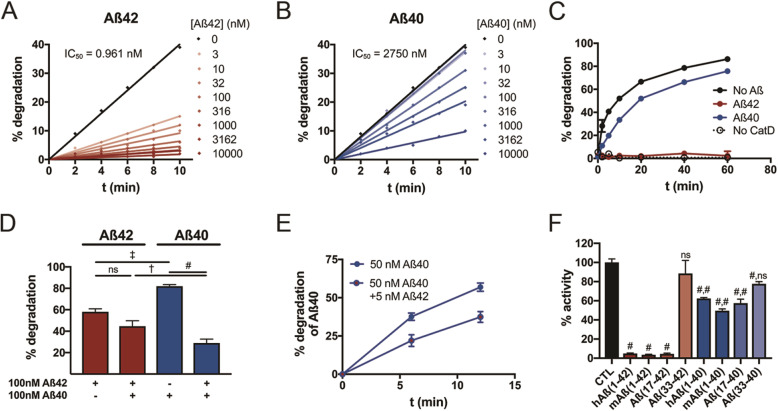
Methods: We quantified Aβ degradation rates and levels of endogenous Aβ42 and Aβ40 in the brains of CatD-null (CatD-KO), heterozygous null (CatD-HET), and wild-type (WT) control mice. CatD-KO mice die by ~ 4 weeks of age, so tissues from younger mice, as well as embryonic neuronal cultures, were investigated. Enzymological assays and surface plasmon resonance were employed to quantify the kinetic parameters (KM, kcat) of CatD-mediated degradation of monomeric human Aβ42 vs. Aβ40, and the degradation of aggregated Aβ42 species was also characterized. Competitive inhibition assays were used to interrogate the relative inhibition of full-length human and mouse Aβ42 and Aβ40, as well as corresponding p3 fragments.
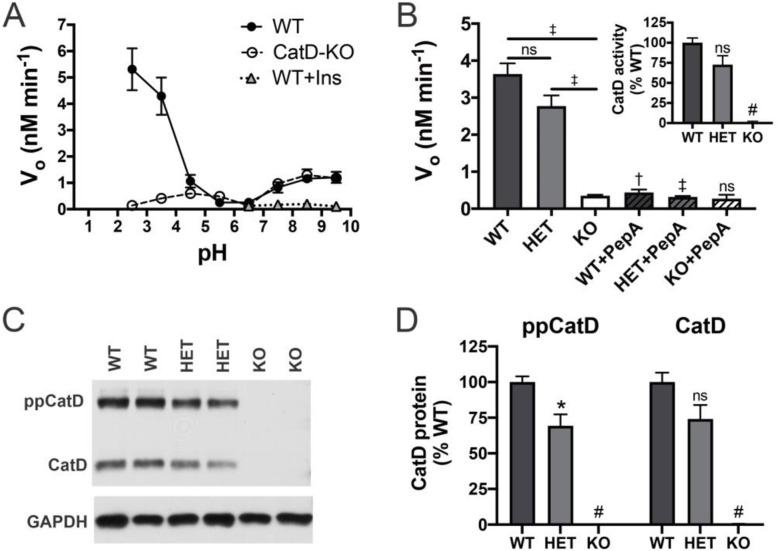
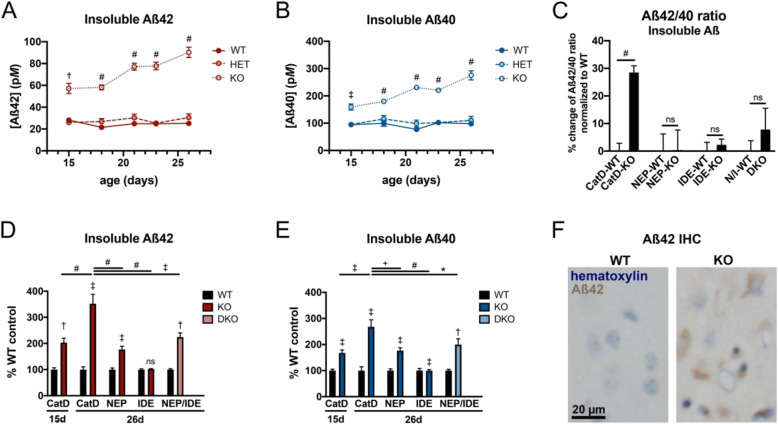
Results: Genetic deletion of CatD resulted in 3- to 4-fold increases in insoluble, endogenous cerebral Aβ42 and Aβ40, exceeding the increases produced by deletion of an insulin-degrading enzyme, neprilysin or both, together with readily detectable intralysosomal deposits of endogenous Aβ42-all by 3 weeks of age. Quite significantly, CatD-KO mice exhibited ~ 30% increases in Aβ42/40 ratios, comparable to those induced by presenilin mutations. Mechanistically, the perturbed Aβ42/40 ratios were attributable to pronounced differences in the kinetics of degradation of Aβ42 vis-à-vis Aβ40. Specifically, Aβ42 shows a low-nanomolar affinity for CatD, along with an exceptionally slow turnover rate that, together, renders Aβ42 a highly potent competitive inhibitor of CatD. Notably, the marked differences in the processing of Aβ42 vs. Aβ40 also extend to p3 fragments ending at positions 42 vs. 40.
Conclusions: Our findings identify CatD as the principal intracellular Aβ-degrading protease identified to date, one that regulates Aβ42/40 ratios via differential degradation of Aβ42 vs. Aβ40. The finding that Aβ42 is a potent competitive inhibitor of CatD suggests a possible mechanistic link between elevations in Aβ42 and downstream pathological sequelae in AD.
Keywords: Alzheimer disease; Amyloid-β protein; Cathepsin D; Lysosomes; Proteostasis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Prominent tauopathy and intracellular β-amyloid accumulation triggered by genetic deletion of cathepsin D: implications for Alzheimer disease pathogenesis.Alzheimers Res Ther. 2024 Apr 4;16(1):70. doi: 10.1186/s13195-024-01443-6. Alzheimers Res Ther. 2024. PMID: 38575959 Free PMC article.
-
Prominent tauopathy and intracellular β-amyloid accumulation triggered by genetic deletion of cathepsin D: Implications for Alzheimer disease pathogenesis.Res Sq [Preprint]. 2023 Oct 23:rs.3.rs-3464352. doi: 10.21203/rs.3.rs-3464352/v1. Res Sq. 2023. Update in: Alzheimers Res Ther. 2024 Apr 4;16(1):70. doi: 10.1186/s13195-024-01443-6. PMID: 37961253 Free PMC article. Updated. Preprint.
-
Cathepsin D: A Candidate Link between Amyloid β-protein and Tauopathy in Alzheimer Disease.J Exp Neurol. 2021;2(1):10-15. J Exp Neurol. 2021. PMID: 33665647 Free PMC article.
-
Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ.J Biol Chem. 2014 Nov 7;289(45):31043-52. doi: 10.1074/jbc.M114.581165. Epub 2014 Sep 19. J Biol Chem. 2014. PMID: 25239621 Free PMC article.
-
Cerebrospinal fluid Abeta40 and Abeta42: natural course and clinical usefulness.Front Biosci. 2002 Apr 1;7:d997-1006. doi: 10.2741/A826. Front Biosci. 2002. PMID: 11897565 Review.
Cited by
-
Topological Dissection of Proteomic Changes Linked to the Limbic Stage of Alzheimer's Disease.Front Immunol. 2021 Oct 12;12:750665. doi: 10.3389/fimmu.2021.750665. eCollection 2021. Front Immunol. 2021. PMID: 34712240 Free PMC article.
-
Autophagy-lysosomal pathway impairment and cathepsin dysregulation in Alzheimer's disease.Front Mol Biosci. 2024 Oct 31;11:1490275. doi: 10.3389/fmolb.2024.1490275. eCollection 2024. Front Mol Biosci. 2024. PMID: 39544403 Free PMC article. Review.
-
Functional genomics identify causal variant underlying the protective CTSH locus for Alzheimer's disease.Neuropsychopharmacology. 2023 Oct;48(11):1555-1566. doi: 10.1038/s41386-023-01542-2. Epub 2023 Feb 4. Neuropsychopharmacology. 2023. PMID: 36739351 Free PMC article.
-
Alzheimer disease.Nat Rev Dis Primers. 2021 May 13;7(1):33. doi: 10.1038/s41572-021-00269-y. Nat Rev Dis Primers. 2021. PMID: 33986301 Free PMC article. Review.
-
The Other Side of Alzheimer's Disease: Influence of Metabolic Disorder Features for Novel Diagnostic Biomarkers.J Pers Med. 2020 Sep 6;10(3):115. doi: 10.3390/jpm10030115. J Pers Med. 2020. PMID: 32899957 Free PMC article. Review.
References
-
- Haass C, Hung AY, Schlossmacher MG, Oltersdorf T, Teplow DB, Selkoe DJ. Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann N Y Acad Sci. 1993;695:109–116. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
