

Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients
- PMID: 32479746
- PMCID: PMC7205692
- DOI: 10.1016/j.cell.2020.05.006
Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients
Abstract
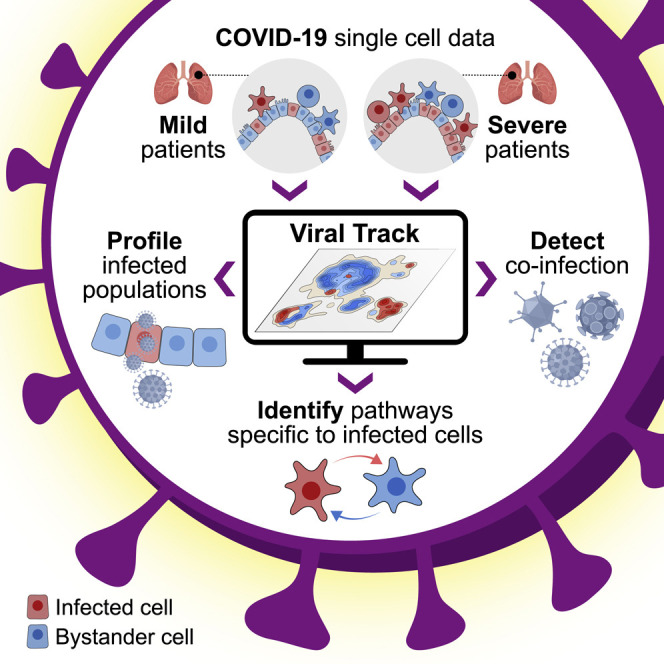
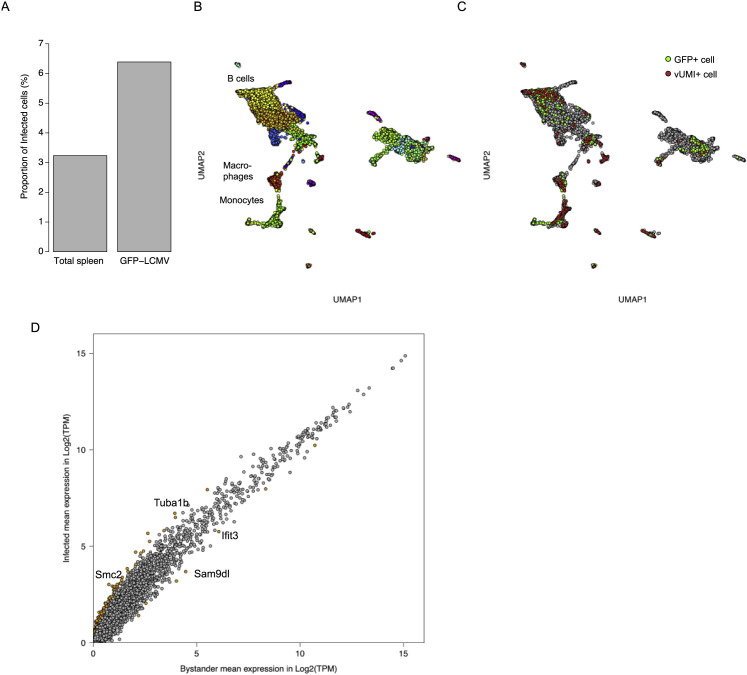
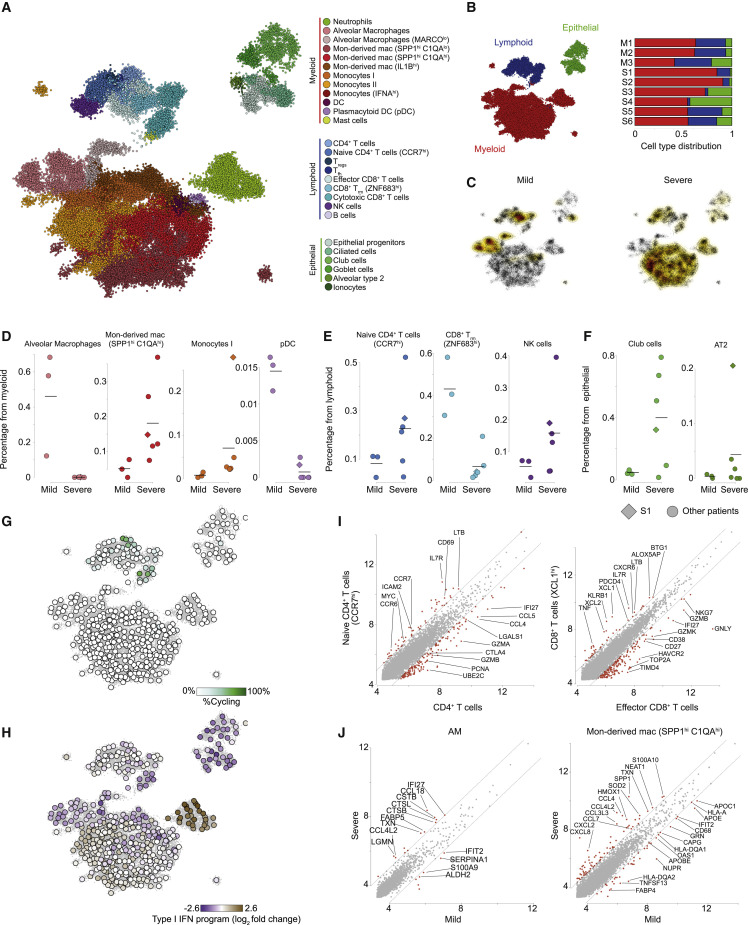
Viruses are a constant threat to global health as highlighted by the current COVID-19 pandemic. Currently, lack of data underlying how the human host interacts with viruses, including the SARS-CoV-2 virus, limits effective therapeutic intervention. We introduce Viral-Track, a computational method that globally scans unmapped single-cell RNA sequencing (scRNA-seq) data for the presence of viral RNA, enabling transcriptional cell sorting of infected versus bystander cells. We demonstrate the sensitivity and specificity of Viral-Track to systematically detect viruses from multiple models of infection, including hepatitis B virus, in an unsupervised manner. Applying Viral-Track to bronchoalveloar-lavage samples from severe and mild COVID-19 patients reveals a dramatic impact of the virus on the immune system of severe patients compared to mild cases. Viral-Track detects an unexpected co-infection of the human metapneumovirus, present mainly in monocytes perturbed in type-I interferon (IFN)-signaling. Viral-Track provides a robust technology for dissecting the mechanisms of viral-infection and pathology.
Keywords: COVID-19; Viral-Track; single-cell RNA-seq; virus host interactions.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures






Similar articles
-
Early Insights into Immune Responses during COVID-19.J Immunol. 2020 Aug 1;205(3):555-564. doi: 10.4049/jimmunol.2000526. Epub 2020 Jun 8. J Immunol. 2020. PMID: 32513850 Review.
-
Primary exposure to SARS-CoV-2 protects against reinfection in rhesus macaques.Science. 2020 Aug 14;369(6505):818-823. doi: 10.1126/science.abc5343. Epub 2020 Jul 2. Science. 2020. PMID: 32616673 Free PMC article.
-
Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19.Nat Med. 2020 Jun;26(6):842-844. doi: 10.1038/s41591-020-0901-9. Epub 2020 May 12. Nat Med. 2020. PMID: 32398875
-
COVID-19: lambda interferon against viral load and hyperinflammation.EMBO Mol Med. 2020 Jun 8;12(6):e12465. doi: 10.15252/emmm.202012465. Epub 2020 May 25. EMBO Mol Med. 2020. PMID: 32333818 Free PMC article.
-
Considering how biological sex impacts immune responses and COVID-19 outcomes.Nat Rev Immunol. 2020 Jul;20(7):442-447. doi: 10.1038/s41577-020-0348-8. Epub 2020 Jun 11. Nat Rev Immunol. 2020. PMID: 32528136 Free PMC article. Review.
Cited by
-
Role of the receptor for advanced glycation end products in the severity of SARS-CoV-2 infection in diabetic patients.Diabetol Int. 2024 Jul 26;15(4):732-744. doi: 10.1007/s13340-024-00746-1. eCollection 2024 Oct. Diabetol Int. 2024. PMID: 39469543 Review.
-
CD68-Negative Histiocytoses with Cardiac Involvement, Associated with COVID-19.Int J Mol Sci. 2024 Sep 19;25(18):10086. doi: 10.3390/ijms251810086. Int J Mol Sci. 2024. PMID: 39337571 Free PMC article.
-
Effect of aberrant fructose metabolism following SARS-CoV-2 infection on colorectal cancer patients' poor prognosis.PLoS Comput Biol. 2024 Sep 27;20(9):e1012412. doi: 10.1371/journal.pcbi.1012412. eCollection 2024 Sep. PLoS Comput Biol. 2024. PMID: 39331675 Free PMC article.
-
Neurological Complications of COVID-19: Unraveling the Pathophysiological Underpinnings and Therapeutic Implications.Viruses. 2024 Jul 24;16(8):1183. doi: 10.3390/v16081183. Viruses. 2024. PMID: 39205157 Free PMC article. Review.
-
COVID-19: from immune response to clinical intervention.Precis Clin Med. 2024 Jul 18;7(3):pbae015. doi: 10.1093/pcmedi/pbae015. eCollection 2024 Sep. Precis Clin Med. 2024. PMID: 39139990 Free PMC article. Review.
References
-
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300.
-
- Blecher-Gonen R., Bost P., Hilligan K.L., David E., Salame T.M., Roussel E., Connor L.M., Mayer J.U., Bahar Halpern K., Tóth B. Single-Cell Analysis of Diverse Pathogen Responses Defines a Molecular Roadmap for Generating Antigen-Specific Immunity. Cell Syst. 2019;8:109–121. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
