

Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases
- PMID: 32371009
- PMCID: PMC7327966
- DOI: 10.1016/j.redox.2020.101506
Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases
Abstract
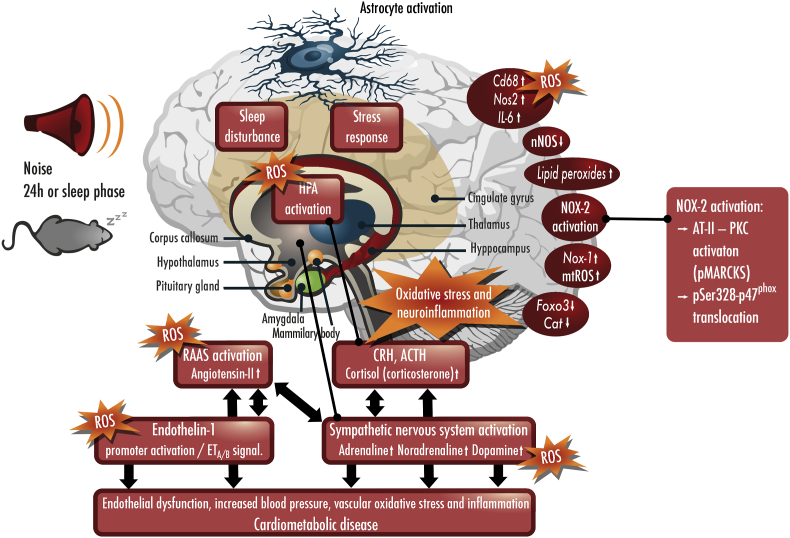
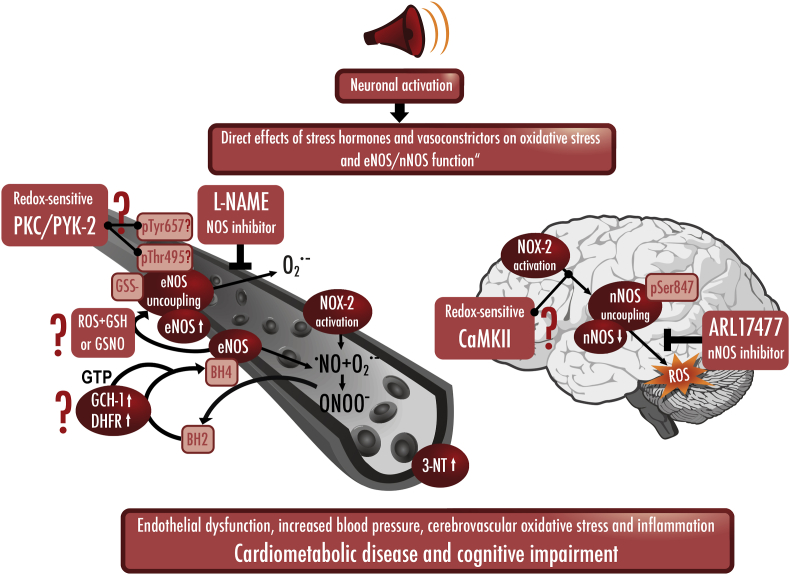
Environmental pollution and non-chemical stressors such as mental stress or traffic noise exposure are increasingly accepted as health risk factors with substantial contribution to chronic noncommunicable diseases (e.g. cardiovascular, metabolic and mental). Whereas the mechanisms of air pollution-mediated adverse health effects are well characterized, the mechanisms of traffic noise exposure are not completely understood, despite convincing clinical and epidemiological evidence for a significant contribution of environmental noise to overall mortality and disability. The initial mechanism of noise-induced cardiovascular, metabolic and mental disease is well defined by the "noise reaction model" and consists of neuronal activation involving the hypothalamic-pituitary-adrenal (HPA) axis as well as the sympathetic nervous system, followed by a classical stress response via cortisol and catecholamines. Stress pathways are initiated by noise-induced annoyance and sleep deprivation/fragmentation. This review highlights the down-stream pathophysiology of noise-induced mental stress, which is based on an induction of inflammation and oxidative stress. We highlight the sources of reactive oxygen species (ROS) involved and the known targets for noise-induced oxidative damage. Part of the review emphasizes noise-triggered uncoupling/dysregulation of endothelial and neuronal nitric oxide synthase (eNOS and nNOS) and its central role for vascular dysfunction.
Keywords: Cardiovascular disease; Environmental risk factors; NOS uncoupling; Oxidative stress; Traffic noise exposure.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures


Similar articles
-
Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation.Eur Heart J. 2018 Oct 7;39(38):3528-3539. doi: 10.1093/eurheartj/ehy333. Eur Heart J. 2018. PMID: 29905797 Free PMC article.
-
Vascular Redox Signaling, Endothelial Nitric Oxide Synthase Uncoupling, and Endothelial Dysfunction in the Setting of Transportation Noise Exposure or Chronic Treatment with Organic Nitrates.Antioxid Redox Signal. 2023 May;38(13-15):1001-1021. doi: 10.1089/ars.2023.0006. Epub 2023 Apr 6. Antioxid Redox Signal. 2023. PMID: 36719770 Free PMC article. Review.
-
Effects of noise on vascular function, oxidative stress, and inflammation: mechanistic insight from studies in mice.Eur Heart J. 2017 Oct 1;38(37):2838-2849. doi: 10.1093/eurheartj/ehx081. Eur Heart J. 2017. PMID: 28329261 Free PMC article.
-
Environmental Noise-Induced Effects on Stress Hormones, Oxidative Stress, and Vascular Dysfunction: Key Factors in the Relationship between Cerebrocardiovascular and Psychological Disorders.Oxid Med Cell Longev. 2019 Nov 11;2019:4623109. doi: 10.1155/2019/4623109. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31814877 Free PMC article. Review.
-
Road traffic noise exposure and its impact on health: evidence from animal and human studies-chronic stress, inflammation, and oxidative stress as key components of the complex downstream pathway underlying noise-induced non-auditory health effects.Environ Sci Pollut Res Int. 2024 Jul;31(34):46820-46839. doi: 10.1007/s11356-024-33973-9. Epub 2024 Jul 8. Environ Sci Pollut Res Int. 2024. PMID: 38977550 Free PMC article. Review.
Cited by
-
Cerebral Blood Flow in Predator Stress-Resilient and -Susceptible Rats and Mechanisms of Resilience.Int J Mol Sci. 2022 Nov 25;23(23):14729. doi: 10.3390/ijms232314729. Int J Mol Sci. 2022. PMID: 36499055 Free PMC article.
-
Schizophrenia and oxidative stress from the perspective of bibliometric analysis.Front Psychiatry. 2023 Feb 27;14:1145409. doi: 10.3389/fpsyt.2023.1145409. eCollection 2023. Front Psychiatry. 2023. PMID: 36923522 Free PMC article.
-
Health position paper and redox perspectives - Disease burden by transportation noise.Redox Biol. 2024 Feb;69:102995. doi: 10.1016/j.redox.2023.102995. Epub 2023 Dec 18. Redox Biol. 2024. PMID: 38142584 Free PMC article. Review.
-
Alpha2-Adrenoblockers Regulate Development of Oxidative Stress and Cognitive Behaviour of Rats under Chronic Acoustic Stress Conditions.Pharmaceuticals (Basel). 2021 Jun 2;14(6):529. doi: 10.3390/ph14060529. Pharmaceuticals (Basel). 2021. PMID: 34199400 Free PMC article.
-
Developmental Exposure to Polychlorinated Biphenyls Prevents Recovery from Noise-Induced Hearing Loss and Disrupts the Functional Organization of the Inferior Colliculus.J Neurosci. 2023 Jun 21;43(25):4580-4597. doi: 10.1523/JNEUROSCI.0030-23.2023. Epub 2023 May 5. J Neurosci. 2023. PMID: 37147134 Free PMC article.
References
-
- Sainani K. Taking on the exposome - bringing bioinformatics tools to the environmental side of the health equation. Biomed. Comput. Rev. 2016;2016:14–21. Fall.
-
- Landrigan P.J., Fuller R., Acosta N.J.R., Adeyi O., Arnold R., Basu N.N., Balde A.B., Bertollini R., Bose-O'Reilly S., Boufford J.I., Breysse P.N., Chiles T., Mahidol C., Coll-Seck A.M., Cropper M.L., Fobil J., Fuster V., Greenstone M., Haines A., Hanrahan D., Hunter D., Khare M., Krupnick A., Lanphear B., Lohani B., Martin K., Mathiasen K.V., McTeer M.A., Murray C.J.L., Ndahimananjara J.D., Perera F., Potocnik J., Preker A.S., Ramesh J., Rockstrom J., Salinas C., Samson L.D., Sandilya K., Sly P.D., Smith K.R., Steiner A., Stewart R.B., Suk W.A., van Schayck O.C.P., Yadama G.N., Yumkella K., Zhong M. The Lancet Commission on pollution and health. Lancet. 2018;391:462–512. doi: 10.1016/S0140-6736(17)32345-0. - DOI - PubMed
-
- WHO Ambient air pollution: a global assessment of exposure and burden of disease. 2016. http://apps.who.int/iris/bitstream/10665/250141/1/9789241511353-eng.pdf?...
-
- WHO report Preventing disease through healthy environments. 2016. https://www.who.int/quantifying_ehimpacts/publications/preventing-diseas...
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
