

Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation
- PMID: 32337344
- PMCID: PMC7164971
- DOI: 10.1212/NXG.0000000000000417
Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation
Abstract
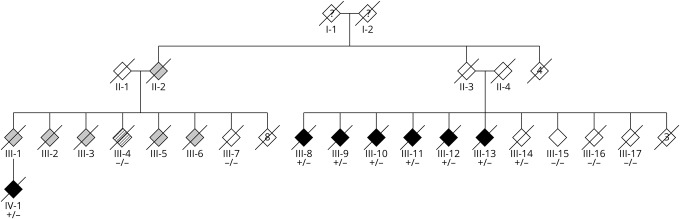
Objective: To describe the clinical and pathologic features of a novel pedigree with heterozygous STUB1 mutation causing SCA48.
Methods: We report a large pedigree of Dutch decent. Clinical and pathologic data were reviewed, and genetic analyses (whole-exome sequencing, whole-genome sequencing, and linkage analysis) were performed on multiple family members.
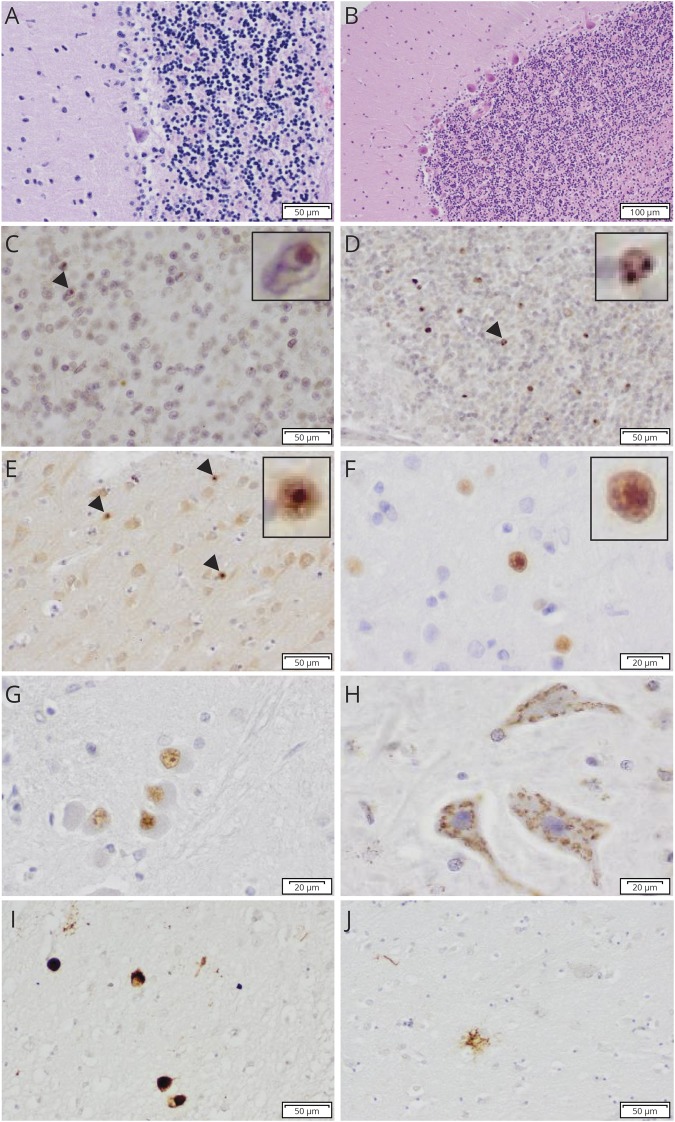
Results: Patients presented with adult-onset gait disturbance (ataxia or parkinsonism), combined with prominent cognitive decline and behavioral changes. Whole-exome sequencing identified a novel heterozygous frameshift variant c.731_732delGC (p.C244Yfs*24) in STUB1 segregating with the disease. This variant was present in a linkage peak on chromosome 16p13.3. Neuropathologic examination of 3 cases revealed a consistent pattern of ubiquitin/p62-positive neuronal inclusions in the cerebellum, neocortex, and brainstem. In addition, tau pathology was present in 1 case.
Conclusions: This study confirms previous findings of heterozygous STUB1 mutations as the cause of SCA48 and highlights its prominent cognitive involvement, besides cerebellar ataxia and movement disorders as cardinal features. The presence of intranuclear inclusions is a pathologic hallmark of the disease. Future studies will provide more insight into its pathologic heterogeneity.
Copyright © 2020 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
Similar articles
-
Novel heterozygous STUB1 gene mutation causes SCA48 in a Hungarian patient.Ideggyogy Sz. 2023 Jan 30;76(1-2):63-72. doi: 10.18071/isz.76.0063. Ideggyogy Sz. 2023. PMID: 36892293 English.
-
Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48).Neurology. 2018 Nov 20;91(21):e1988-e1998. doi: 10.1212/WNL.0000000000006550. Epub 2018 Oct 31. Neurology. 2018. PMID: 30381368
-
Clinical and functional characterization of a novel STUB1 frameshift mutation in autosomal dominant spinocerebellar ataxia type 48 (SCA48).J Biomed Sci. 2021 Sep 26;28(1):65. doi: 10.1186/s12929-021-00763-1. J Biomed Sci. 2021. PMID: 34565360 Free PMC article.
-
Spinocerebellar ataxia type 48: last but not least.Neurol Sci. 2020 Sep;41(9):2423-2432. doi: 10.1007/s10072-020-04408-3. Epub 2020 Apr 27. Neurol Sci. 2020. PMID: 32342324 Review.
-
Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias.Hum Genome Var. 2014 Oct 23;1:14018. doi: 10.1038/hgv.2014.18. eCollection 2014. Hum Genome Var. 2014. PMID: 27081508 Free PMC article. Review.
Cited by
-
The molecular basis of spinocerebellar ataxia type 48 caused by a de novo mutation in the ubiquitin ligase CHIP.J Biol Chem. 2022 May;298(5):101899. doi: 10.1016/j.jbc.2022.101899. Epub 2022 Apr 7. J Biol Chem. 2022. PMID: 35398354 Free PMC article.
-
A Severe Dementia Syndrome Caused by Intron Retention and Cryptic Splice Site Activation in STUB1 and Exacerbated by TBP Repeat Expansions.Front Mol Neurosci. 2022 Apr 14;15:878236. doi: 10.3389/fnmol.2022.878236. eCollection 2022. Front Mol Neurosci. 2022. PMID: 35493319 Free PMC article.
-
A de novo STUB1 variant associated with an early adult-onset multisystemic ataxia phenotype.J Neurol. 2021 Oct;268(10):3845-3851. doi: 10.1007/s00415-021-10524-7. Epub 2021 Apr 3. J Neurol. 2021. PMID: 33811518 Free PMC article.
-
The chaperone-assisted selective autophagy complex dynamics and dysfunctions.Autophagy. 2023 Jun;19(6):1619-1641. doi: 10.1080/15548627.2022.2160564. Epub 2023 Jan 3. Autophagy. 2023. PMID: 36594740 Free PMC article.
-
Clinical and genetic analyses of a Swedish patient series diagnosed with ataxia.J Neurol. 2024 Jan;271(1):526-542. doi: 10.1007/s00415-023-11990-x. Epub 2023 Oct 3. J Neurol. 2024. PMID: 37787810 Free PMC article.
References
-
- Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers 2019;5:24. - PubMed
-
- Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012;124:1–21. - PubMed
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. - PubMed
-
- Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42:174–183. - PubMed
-
- Synofzik M, Nemeth AH. Recessive ataxias. Handb Clin Neurol 2018;155:73–89. - PubMed
LinkOut - more resources
Full Text Sources