

A Pan-plant Protein Complex Map Reveals Deep Conservation and Novel Assemblies
- PMID: 32191846
- PMCID: PMC7297045
- DOI: 10.1016/j.cell.2020.02.049
A Pan-plant Protein Complex Map Reveals Deep Conservation and Novel Assemblies
Abstract
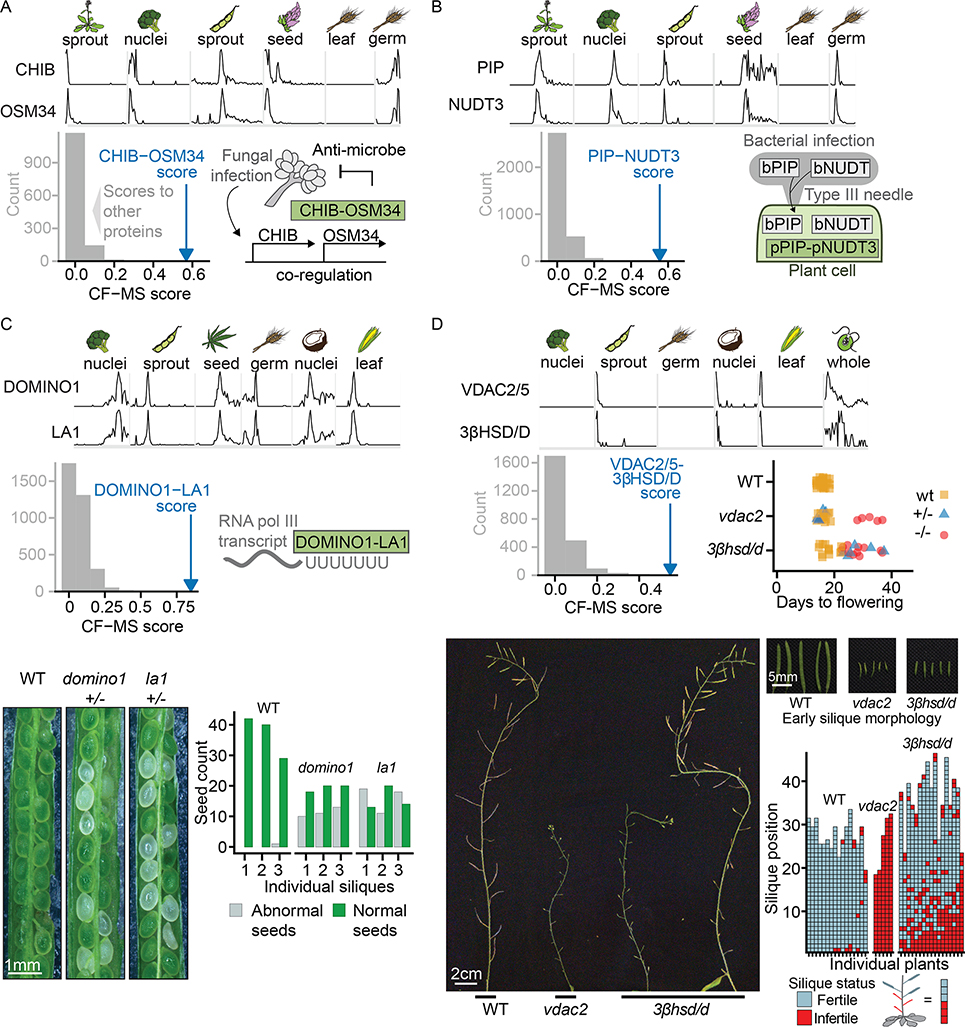
Plants are foundational for global ecological and economic systems, but most plant proteins remain uncharacterized. Protein interaction networks often suggest protein functions and open new avenues to characterize genes and proteins. We therefore systematically determined protein complexes from 13 plant species of scientific and agricultural importance, greatly expanding the known repertoire of stable protein complexes in plants. By using co-fractionation mass spectrometry, we recovered known complexes, confirmed complexes predicted to occur in plants, and identified previously unknown interactions conserved over 1.1 billion years of green plant evolution. Several novel complexes are involved in vernalization and pathogen defense, traits critical for agriculture. We also observed plant analogs of animal complexes with distinct molecular assemblies, including a megadalton-scale tRNA multi-synthetase complex. The resulting map offers a cross-species view of conserved, stable protein assemblies shared across plant cells and provides a mechanistic, biochemical framework for interpreting plant genetics and mutant phenotypes.
Keywords: co-fractionation mass spectrometry (CF-MS); comparative proteomics; cross-linking mass spectrometry (CL-MS); evolution; interaction-to-phenotype; pathogen defense; plants; protein complexes; protein interactions.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
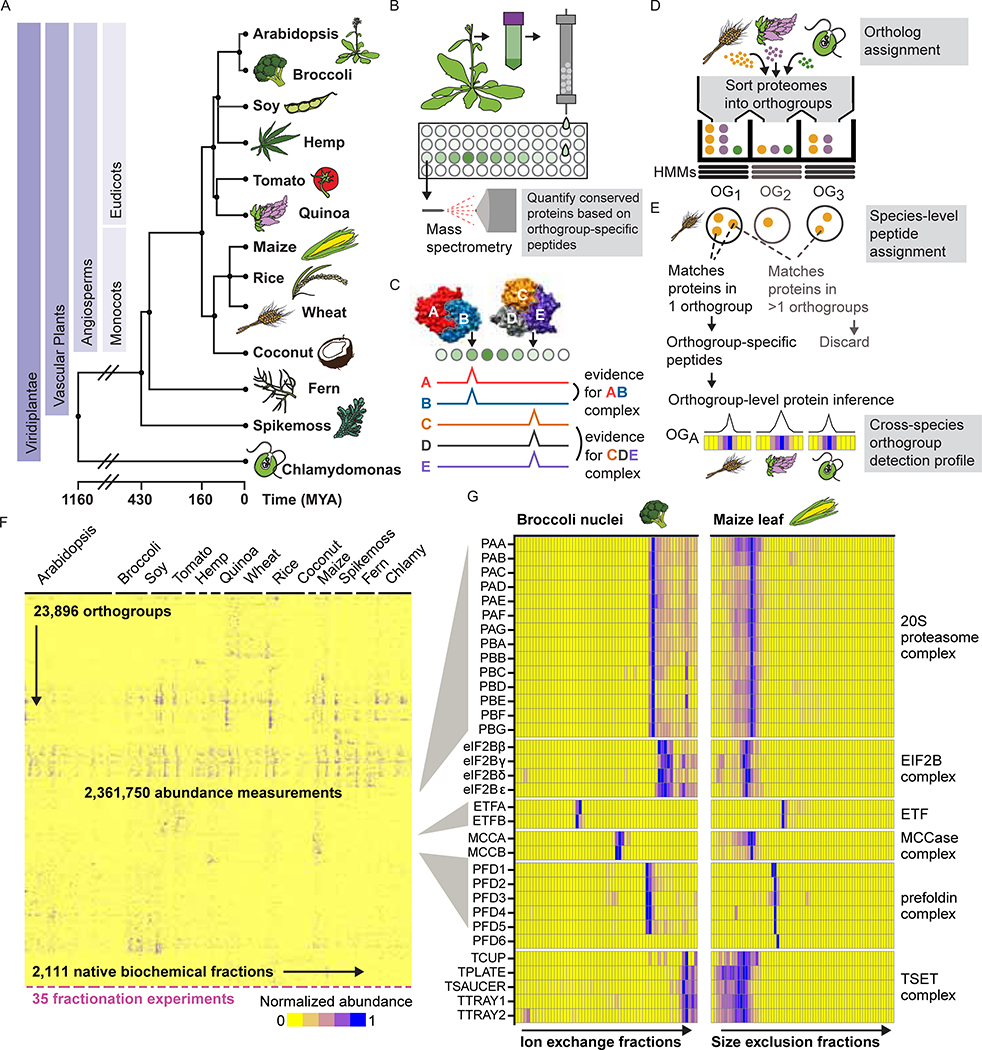

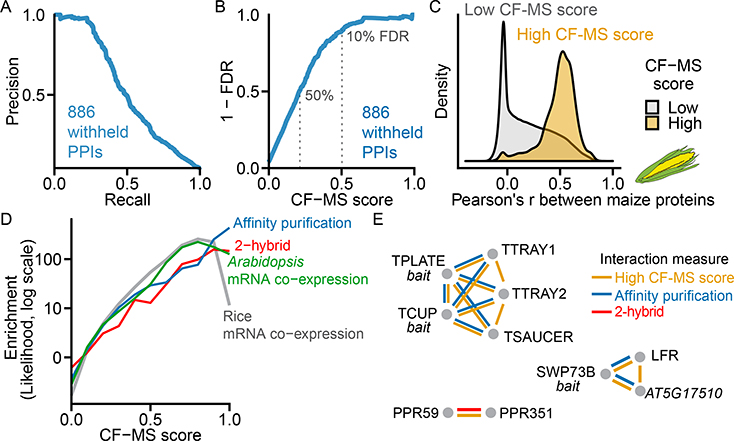
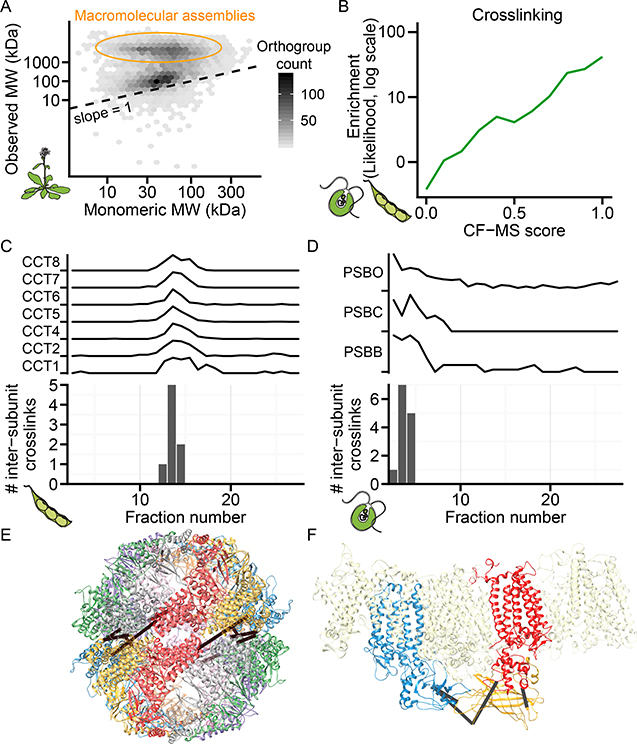
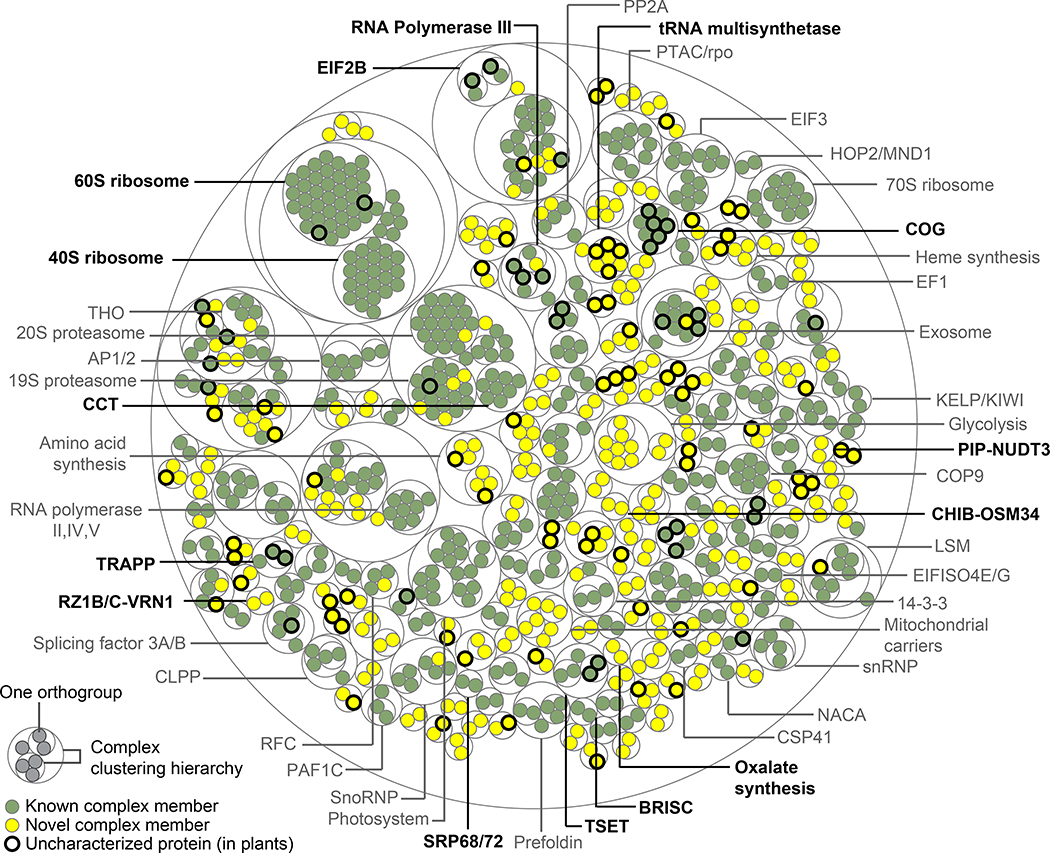

Comment in
-
An Abundance and Interaction Encyclopedia of Plant Protein Function.Trends Plant Sci. 2020 Jul;25(7):627-630. doi: 10.1016/j.tplants.2020.04.006. Epub 2020 Apr 26. Trends Plant Sci. 2020. PMID: 32349926
Similar articles
-
In Vivo Cross-Linking to Analyze Transient Protein-Protein Interactions.Methods Mol Biol. 2020;2139:273-287. doi: 10.1007/978-1-0716-0528-8_20. Methods Mol Biol. 2020. PMID: 32462593
-
Trends in co-fractionation mass spectrometry: A new gold-standard in global protein interaction network discovery.Curr Opin Struct Biol. 2024 Oct;88:102880. doi: 10.1016/j.sbi.2024.102880. Epub 2024 Jul 11. Curr Opin Struct Biol. 2024. PMID: 38996623 Review.
-
Spt-Ada-Gcn5-Acetyltransferase (SAGA) Complex in Plants: Genome Wide Identification, Evolutionary Conservation and Functional Determination.PLoS One. 2015 Aug 11;10(8):e0134709. doi: 10.1371/journal.pone.0134709. eCollection 2015. PLoS One. 2015. PMID: 26263547 Free PMC article.
-
Unravelling plant molecular machineries through affinity purification coupled to mass spectrometry.Curr Opin Plant Biol. 2015 Apr;24:1-9. doi: 10.1016/j.pbi.2015.01.001. Epub 2015 Jan 17. Curr Opin Plant Biol. 2015. PMID: 25603557 Review.
-
Computational framework for analysis of prey-prey associations in interaction proteomics identifies novel human protein-protein interactions and networks.J Proteome Res. 2012 Sep 7;11(9):4476-87. doi: 10.1021/pr300227y. Epub 2012 Aug 21. J Proteome Res. 2012. PMID: 22845868 Free PMC article.
Cited by
-
Current status of the multinational Arabidopsis community.Plant Direct. 2020 Aug 2;4(7):e00248. doi: 10.1002/pld3.248. eCollection 2020 Jul. Plant Direct. 2020. PMID: 32775952 Free PMC article.
-
Glucose-6-P/phosphate translocator2 mediates the phosphoglucose-isomerase1-independent response to microbial volatiles.Plant Physiol. 2022 Nov 28;190(4):2137-2154. doi: 10.1093/plphys/kiac433. Plant Physiol. 2022. PMID: 36111879 Free PMC article.
-
A systematic, label-free method for identifying RNA-associated proteins in vivo provides insights into vertebrate ciliary beating machinery.Dev Biol. 2020 Nov 1;467(1-2):108-117. doi: 10.1016/j.ydbio.2020.08.008. Epub 2020 Sep 6. Dev Biol. 2020. PMID: 32898505 Free PMC article.
-
Protein-protein interactions in plant antioxidant defense.Front Plant Sci. 2022 Dec 14;13:1035573. doi: 10.3389/fpls.2022.1035573. eCollection 2022. Front Plant Sci. 2022. PMID: 36589041 Free PMC article. Review.
-
Defects in autophagy lead to selective in vivo changes in turnover of cytosolic and organelle proteins in Arabidopsis.Plant Cell. 2022 Sep 27;34(10):3936-3960. doi: 10.1093/plcell/koac185. Plant Cell. 2022. PMID: 35766863 Free PMC article.
References
-
- Abdrakhmanova A, Dobrynin K, Zwicker K, Kerscher S, and Brandt U (2005). Functional sulfurtransferase is associated with mitochondrial complex I from Yarrowia lipolytica, but is not required for assembly of its iron–sulfur clusters. FEBS Lett. 579, 6781–6785. - PubMed
-
- Adamiec M, Ciesielska M, Zalaś P, and Luciński R (2017). Arabidopsis thaliana intramembrane proteases. Acta Physiol. Plant. 39, 146.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
