

The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy
- PMID: 32144382
- PMCID: PMC7370224
- DOI: 10.1038/s41418-020-0514-3
The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy
Erratum in
-
Correction: The lipogenic LXR-SREBF1 signaling pathway controls cancer cell DNA repair and apoptosis and is a vulnerable point of malignant tumors for cancer therapy.Cell Death Differ. 2020 Sep;27(9):2746. doi: 10.1038/s41418-020-0534-z. Cell Death Differ. 2020. PMID: 32249819 Free PMC article.
Abstract
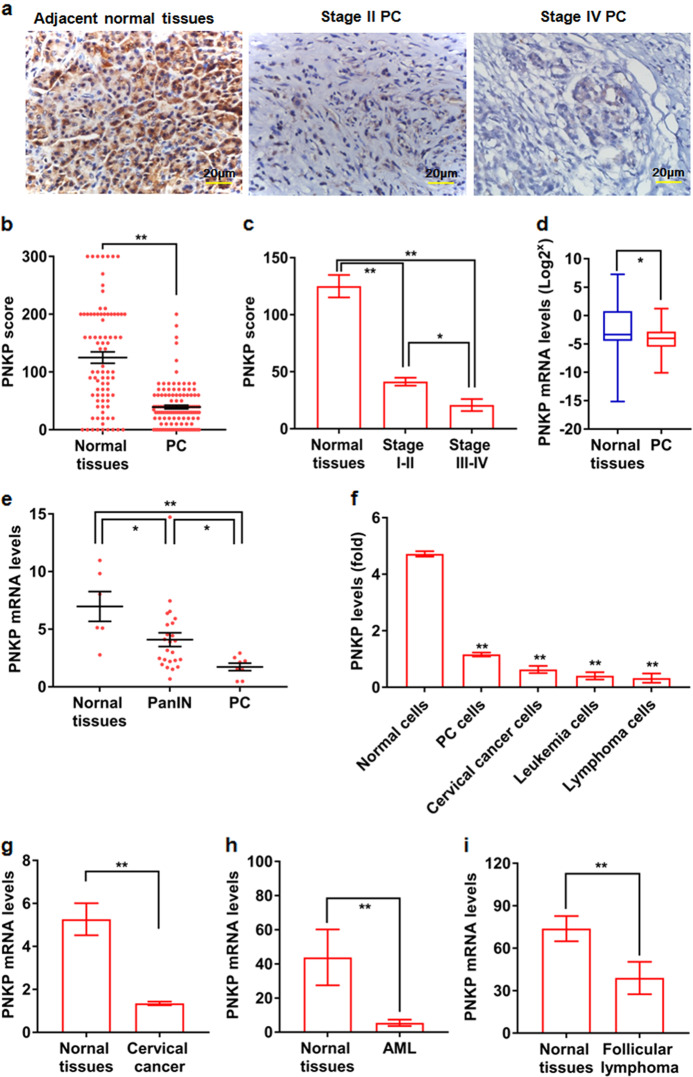
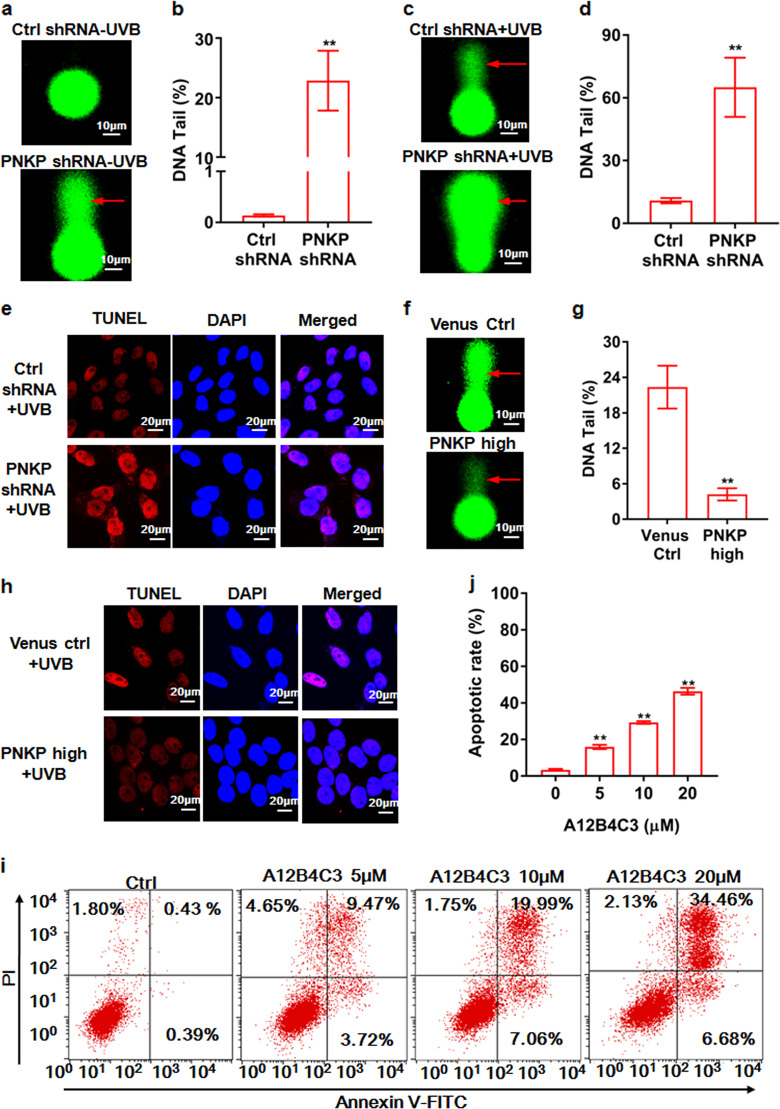
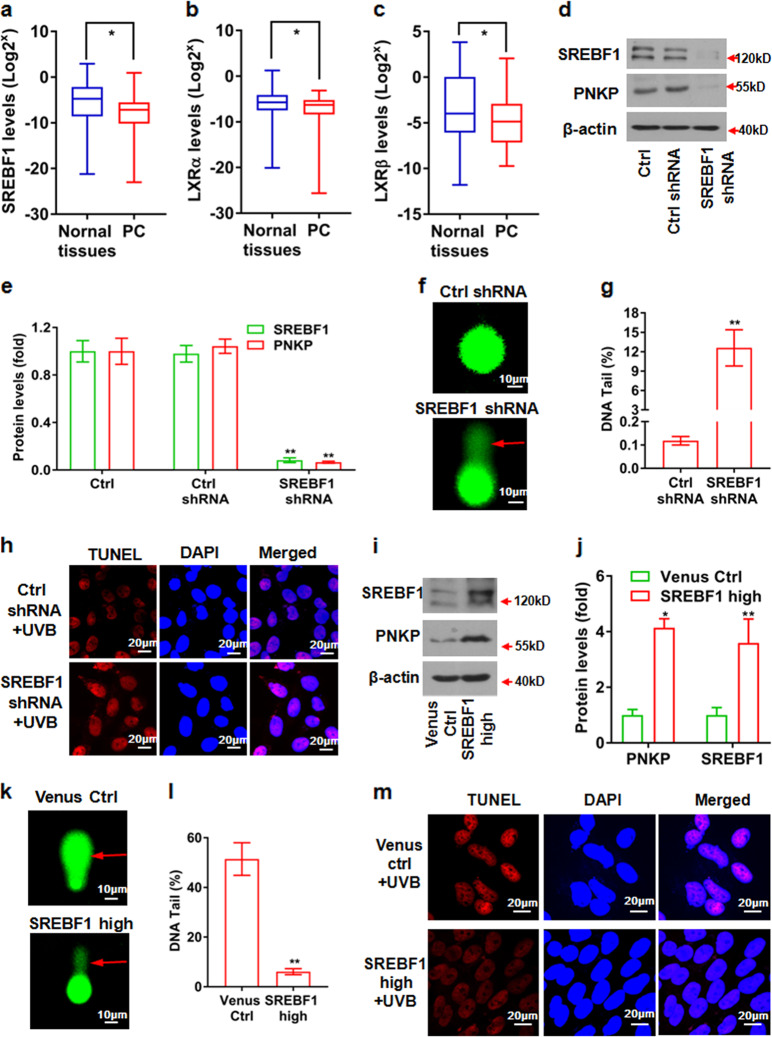
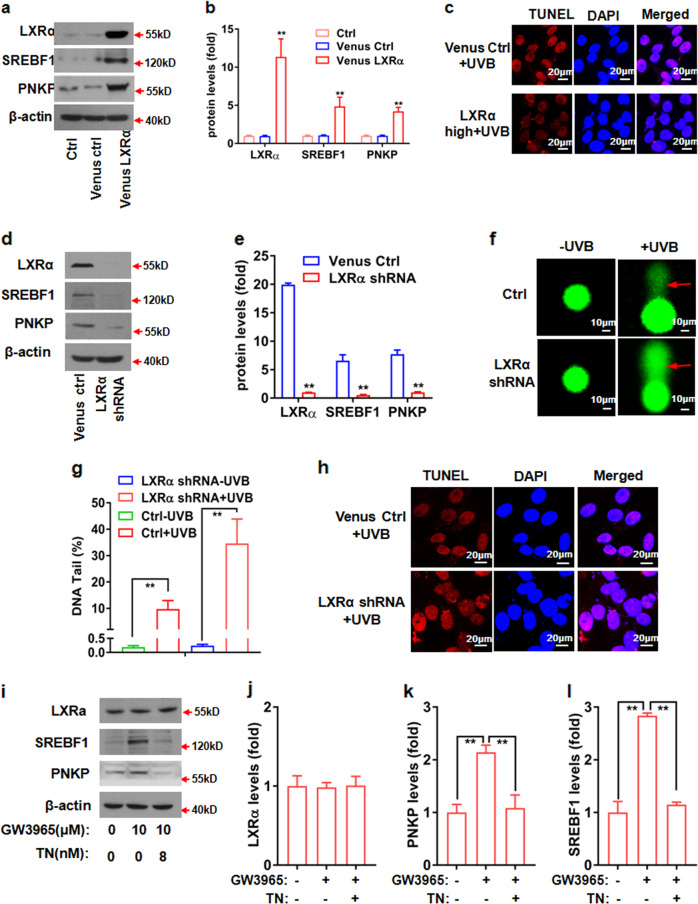
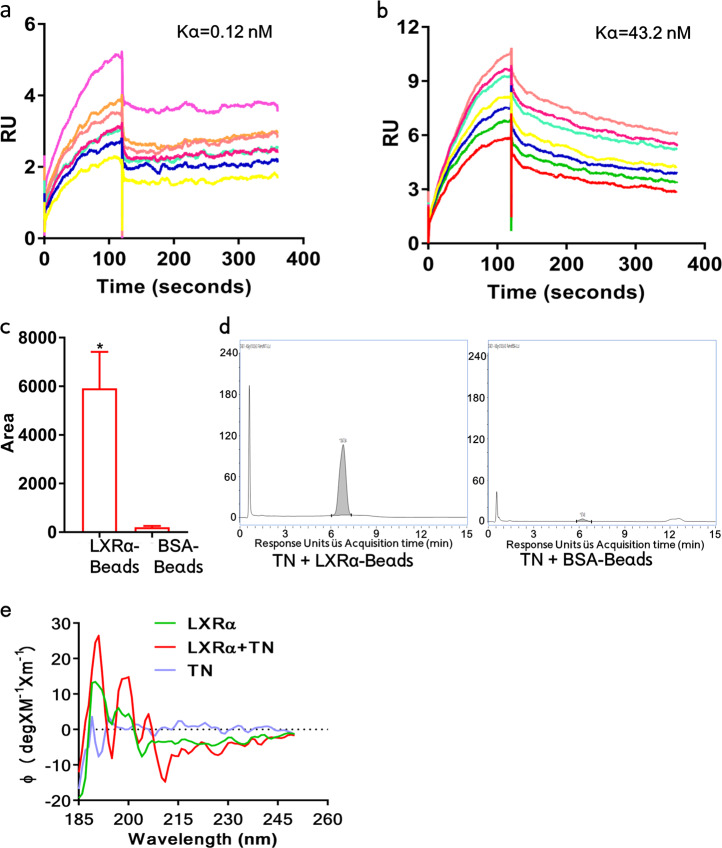
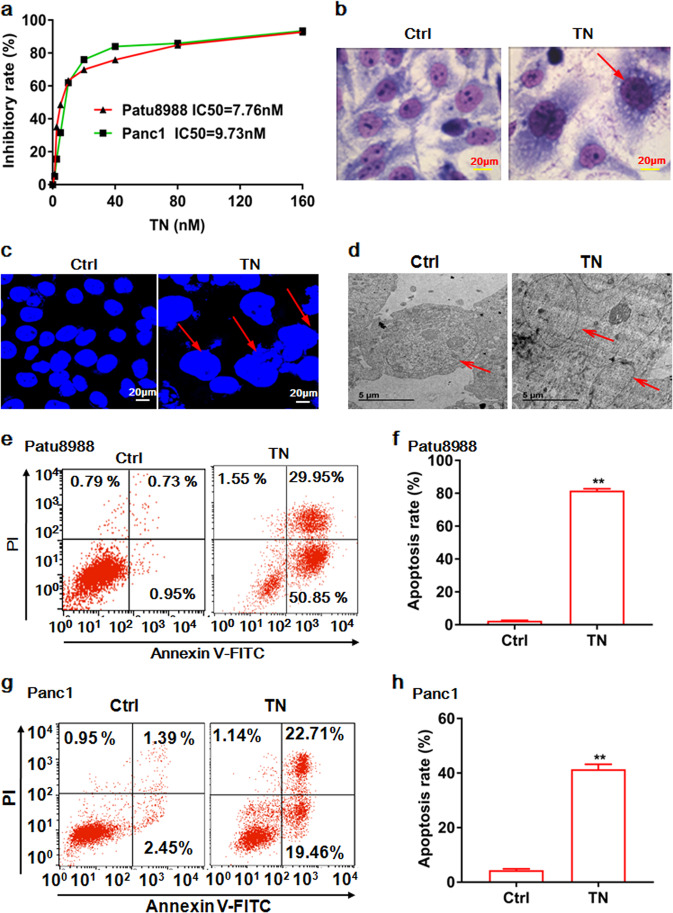
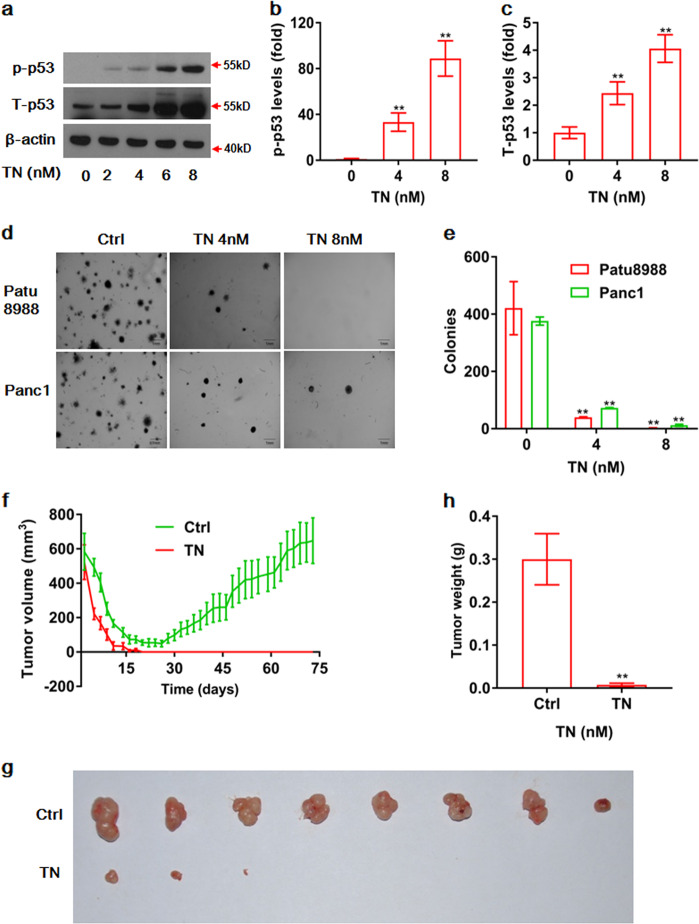
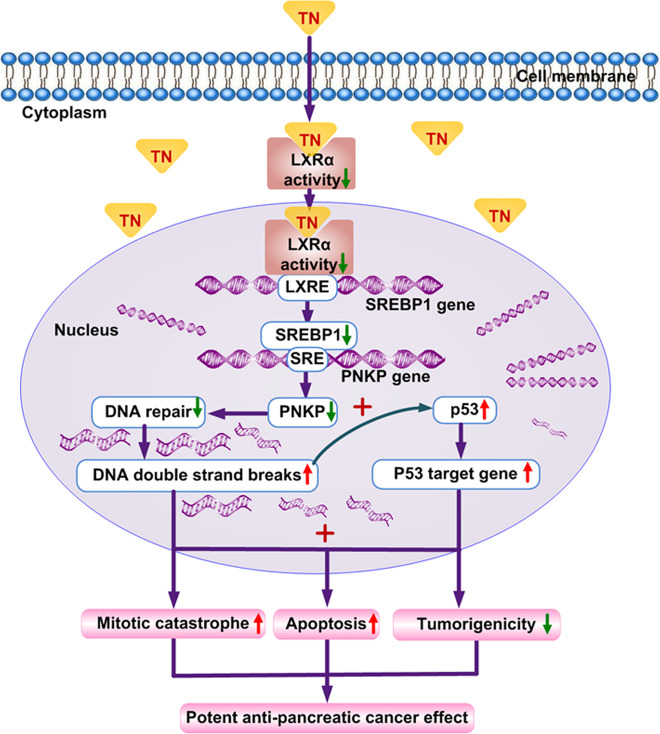
Cancer cells are defective in DNA repair, so they experience increased DNA strand breaks, genome instability, gene mutagenesis, and tumorigenicity; however, multiple classic DNA repair genes and pathways are strongly activated in malignant tumor cells to compensate for the DNA repair deficiency and gain an apoptosis resistance. The mechanisms underlying this phenomenon in cancer are unclear. We speculate that a key DNA repair gene or signaling pathway in cancer has not yet been recognized. Here, we show that the lipogenic liver X receptor (LXR)-sterol response element binding factor-1 (SREBF1) axis controls the transcription of a key DNA repair gene polynucleotide kinase/phosphatase (PNKP), thereby governing cancer cell DNA repair and apoptosis. Notably, the PNKP levels were significantly reduced in 95% of human pancreatic cancer (PC) patients, particularly deep reduction for sixfold in all of the advanced-stage PC cases. PNKP is also deficient in three other types of cancer that we examined. In addition, the expression of LXRs and SREBF1 was significantly reduced in the tumor tissues from human PC patients compared with the adjacent normal tissues. The newly identified LXR-SREBF1-PNKP signaling pathway is deficient in PC, and the defect in the pathway contributes to the DNA repair deficiency in the cancer. Strikingly, further diminution of the vulnerable LXR-SREBF1-PNKP signaling pathway using a small molecule triptonide, a new LXR antagonist identified in this investigation, at a concentration of 8 nM robustly activated tumor-suppressor p53 and readily elevated cancer cell DNA strand breaks over an apoptotic threshold, and selectively induced PC cell apoptosis, resulting in almost complete elimination of tumors in xenograft mice without obvious complications. Our findings provide new insight into DNA repair and apoptosis in cancer, and offer a new platform for developing novel anticancer therapeutics.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Synthetic lethal targeting of PTEN-deficient cancer cells using selective disruption of polynucleotide kinase/phosphatase.Mol Cancer Ther. 2013 Oct;12(10):2135-44. doi: 10.1158/1535-7163.MCT-12-1093. Epub 2013 Jul 24. Mol Cancer Ther. 2013. PMID: 23883586 Free PMC article.
-
Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage.Nucleic Acids Res. 2011 Nov;39(21):9224-37. doi: 10.1093/nar/gkr647. Epub 2011 Aug 8. Nucleic Acids Res. 2011. PMID: 21824916 Free PMC article.
-
Pathological mutations in PNKP trigger defects in DNA single-strand break repair but not DNA double-strand break repair.Nucleic Acids Res. 2020 Jul 9;48(12):6672-6684. doi: 10.1093/nar/gkaa489. Nucleic Acids Res. 2020. PMID: 32504494 Free PMC article.
-
Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair.Trends Biochem Sci. 2011 May;36(5):262-71. doi: 10.1016/j.tibs.2011.01.006. Epub 2011 Feb 25. Trends Biochem Sci. 2011. PMID: 21353781 Free PMC article. Review.
-
Polynucleotide kinase-phosphatase (PNKP) mutations and neurologic disease.Mech Ageing Dev. 2017 Jan;161(Pt A):121-129. doi: 10.1016/j.mad.2016.04.009. Epub 2016 Apr 26. Mech Ageing Dev. 2017. PMID: 27125728 Free PMC article. Review.
Cited by
-
Enhanced cancer cell proliferation and aggressive phenotype counterbalance in breast cancer with high BRCA1 gene expression.Breast Cancer Res Treat. 2024 Nov;208(2):321-331. doi: 10.1007/s10549-024-07421-8. Epub 2024 Jul 7. Breast Cancer Res Treat. 2024. PMID: 38972017 Free PMC article.
-
Liver X Receptors (LXRs) in cancer-an Eagle's view on molecular insights and therapeutic opportunities.Front Cell Dev Biol. 2024 Mar 14;12:1386102. doi: 10.3389/fcell.2024.1386102. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 38550382 Free PMC article. Review.
-
Lipid Metabolism as a Potential Target of Liver Cancer.J Hepatocell Carcinoma. 2024 Feb 14;11:327-346. doi: 10.2147/JHC.S450423. eCollection 2024. J Hepatocell Carcinoma. 2024. PMID: 38375401 Free PMC article. Review.
-
Emerging Insights into Liver X Receptor α in the Tumorigenesis and Therapeutics of Human Cancers.Biomolecules. 2023 Jul 28;13(8):1184. doi: 10.3390/biom13081184. Biomolecules. 2023. PMID: 37627249 Free PMC article. Review.
-
Triptonide effectively inhibits triple-negative breast cancer metastasis through concurrent degradation of Twist1 and Notch1 oncoproteins.Breast Cancer Res. 2021 Dec 18;23(1):116. doi: 10.1186/s13058-021-01488-7. Breast Cancer Res. 2021. PMID: 34922602 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
