

Impact of HMGB1, RAGE, and TLR4 in Alzheimer's Disease (AD): From Risk Factors to Therapeutic Targeting
- PMID: 32046119
- PMCID: PMC7072620
- DOI: 10.3390/cells9020383
Impact of HMGB1, RAGE, and TLR4 in Alzheimer's Disease (AD): From Risk Factors to Therapeutic Targeting
Abstract
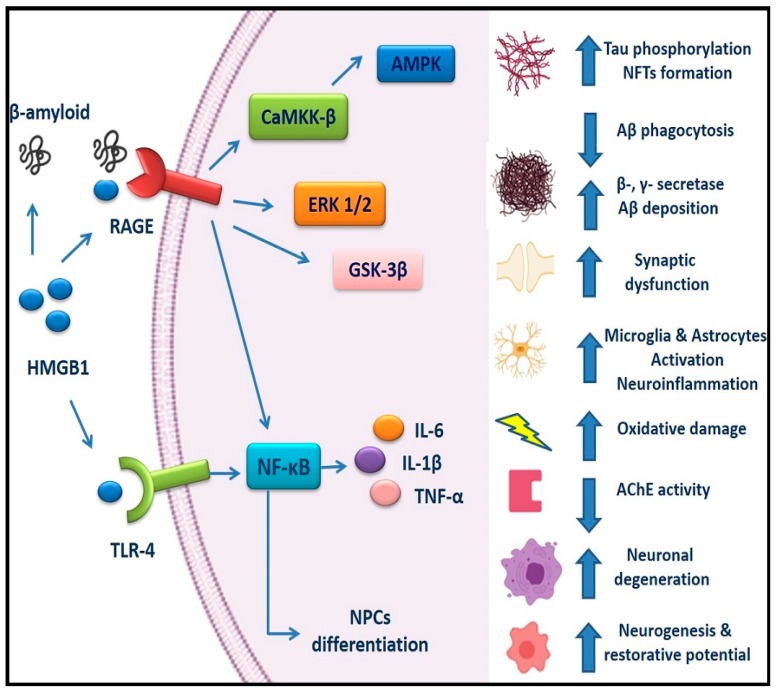
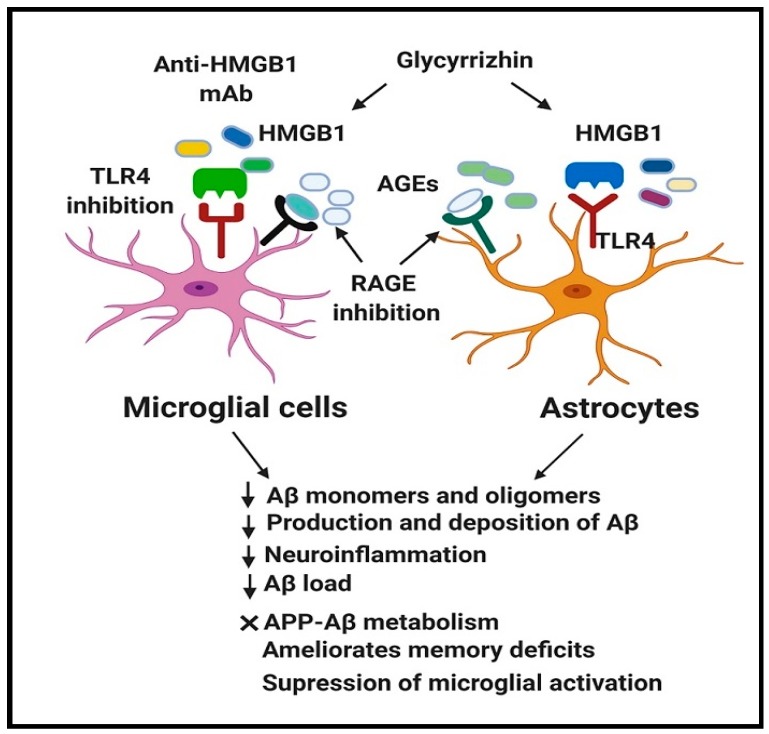
Alzheimer's disease (AD) is a devastating neurodegenerative disorder and a leading cause of dementia, with accumulation of amyloid-beta (Aβ) and neurofibrillary tangles (NFTs) as defining pathological features. AD presents a serious global health concern with no cure to date, reflecting the complexity of its pathogenesis. Recent evidence indicates that neuroinflammation serves as the link between amyloid deposition, Tau pathology, and neurodegeneration. The high mobility group box 1 (HMGB1) protein, an initiator and activator of neuroinflammatory responses, has been involved in the pathogenesis of neurodegenerative diseases, including AD. HMGB1 is a typical damage-associated molecular pattern (DAMP) protein that exerts its biological activity mainly through binding to the receptor for advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4). RAGE and TLR4 are key components of the innate immune system that both bind to HMGB1. Targeting of HMGB1, RAGE, and TLR4 in experimental AD models has demonstrated beneficial effects in halting AD progression by suppressing neuroinflammation, reducing Aβ load and production, improving spatial learning, and inhibiting microglial stimulation. Herein, we discuss the contribution of HMGB1 and its receptor signaling in neuroinflammation and AD pathogenesis, providing evidence of its beneficial effects upon therapeutic targeting.
Keywords: Alzheimer’s disease; HMGB1; Neuroinflammation; RAGE; TLR4.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
AGE-RAGE stress: a changing landscape in pathology and treatment of Alzheimer's disease.Mol Cell Biochem. 2019 Sep;459(1-2):95-112. doi: 10.1007/s11010-019-03553-4. Epub 2019 May 11. Mol Cell Biochem. 2019. PMID: 31079281 Review.
-
Implication of HMGB1 signaling pathways in Amyotrophic lateral sclerosis (ALS): From molecular mechanisms to pre-clinical results.Pharmacol Res. 2020 Jun;156:104792. doi: 10.1016/j.phrs.2020.104792. Epub 2020 Apr 8. Pharmacol Res. 2020. PMID: 32278047 Review.
-
Curcumin improves memory deficits by inhibiting HMGB1-RAGE/TLR4-NF-κB signalling pathway in APPswe/PS1dE9 transgenic mice hippocampus.J Cell Mol Med. 2021 Sep;25(18):8947-8956. doi: 10.1111/jcmm.16855. Epub 2021 Aug 18. J Cell Mol Med. 2021. PMID: 34405526 Free PMC article.
-
Electroacupuncture Improves Blood-Brain Barrier and Hippocampal Neuroinflammation in SAMP8 Mice by Inhibiting HMGB1/TLR4 and RAGE/NADPH Signaling Pathways.Chin J Integr Med. 2023 May;29(5):448-458. doi: 10.1007/s11655-023-3592-5. Epub 2023 Jan 7. Chin J Integr Med. 2023. PMID: 36609953
-
High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells.Am J Physiol Lung Cell Mol Physiol. 2015 Dec 1;309(11):L1354-66. doi: 10.1152/ajplung.00054.2015. Epub 2015 Oct 2. Am J Physiol Lung Cell Mol Physiol. 2015. PMID: 26432865
Cited by
-
Advanced Glycation End Products-Induced Alzheimer's Disease and Its Novel Therapeutic Approaches: A Comprehensive Review.Cureus. 2024 May 30;16(5):e61373. doi: 10.7759/cureus.61373. eCollection 2024 May. Cureus. 2024. PMID: 38947632 Free PMC article. Review.
-
Amyloid Proteins and Peripheral Neuropathy.Cells. 2020 Jun 26;9(6):1553. doi: 10.3390/cells9061553. Cells. 2020. PMID: 32604774 Free PMC article. Review.
-
Non-toxic nature of chebulinic acid on biochemical, hematological and histopathological analysis in normal Sprague Dawley rats.Toxicol Res. 2021 Apr 24;38(2):159-174. doi: 10.1007/s43188-021-00092-3. eCollection 2022 Apr. Toxicol Res. 2021. PMID: 35419271 Free PMC article.
-
High-Mobility Group Box-1 and Its Potential Role in Perioperative Neurocognitive Disorders.Cells. 2021 Sep 28;10(10):2582. doi: 10.3390/cells10102582. Cells. 2021. PMID: 34685561 Free PMC article. Review.
-
Microglia and Astrocytes in Alzheimer's Disease: Significance and Summary of Recent Advances.Aging Dis. 2024 Aug 1;15(4):1537-1564. doi: 10.14336/AD.2023.0907. Aging Dis. 2024. PMID: 37815901 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
