

Pan-cancer analysis of whole genomes
- PMID: 32025007
- PMCID: PMC7025898
- DOI: 10.1038/s41586-020-1969-6
Pan-cancer analysis of whole genomes
Erratum in
-
Author Correction: Pan-cancer analysis of whole genomes.Nature. 2023 Feb;614(7948):E39. doi: 10.1038/s41586-022-05598-w. Nature. 2023. PMID: 36697834 Free PMC article. No abstract available.
Abstract
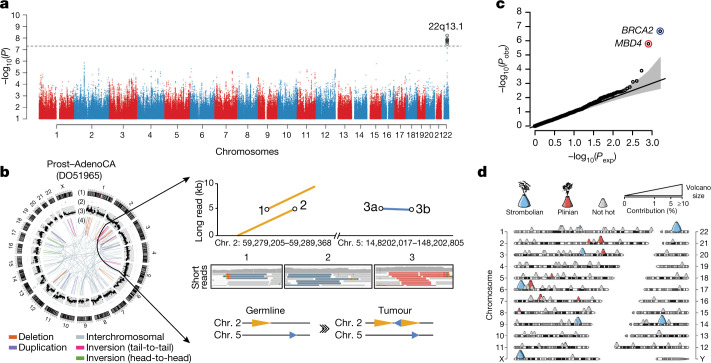
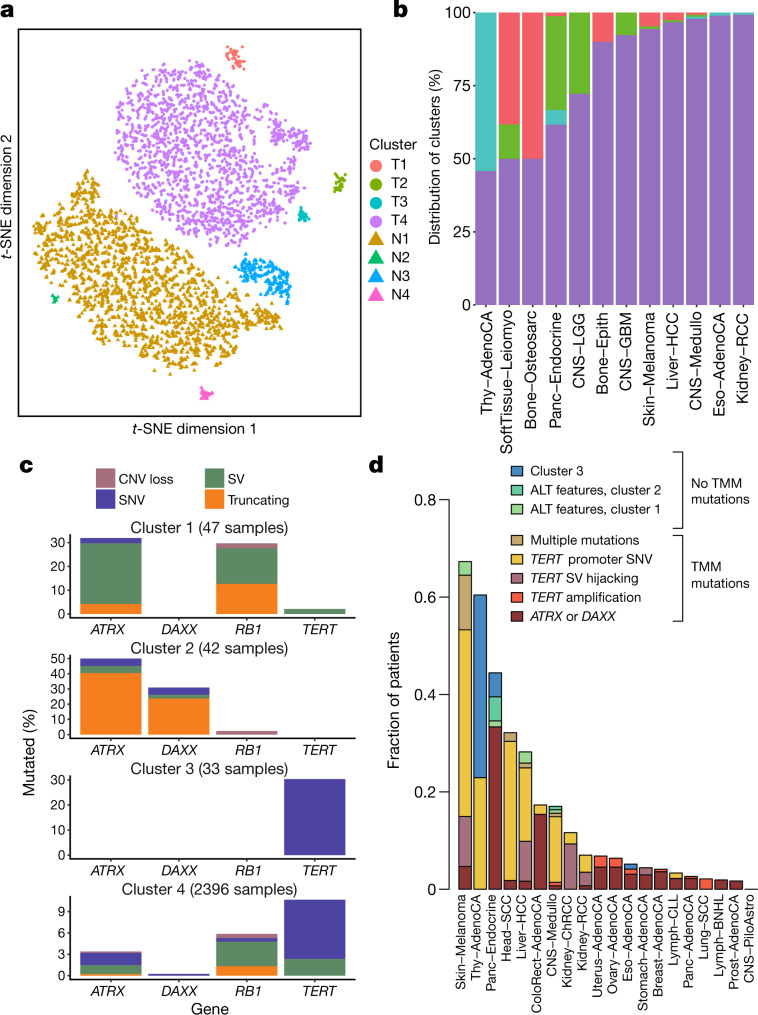
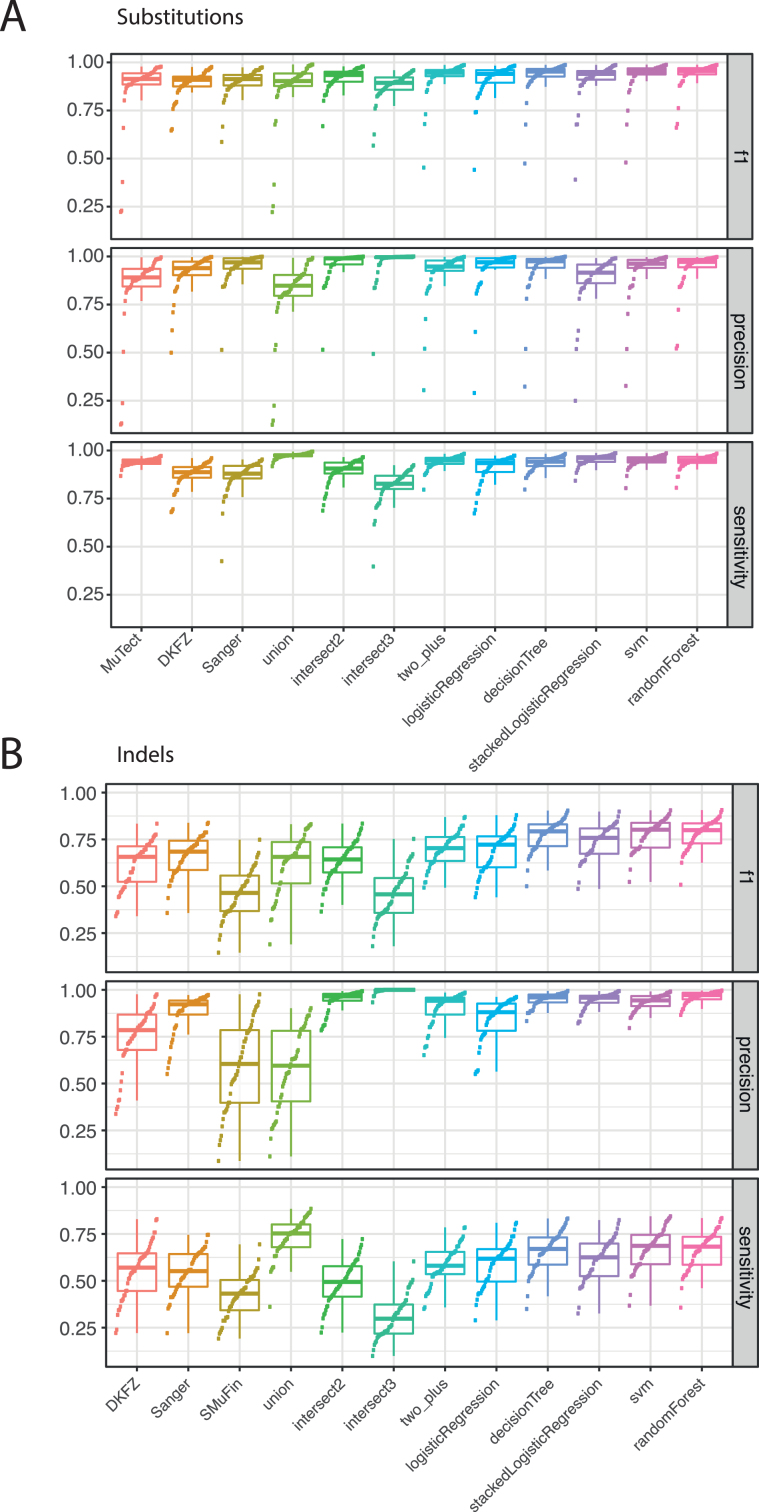
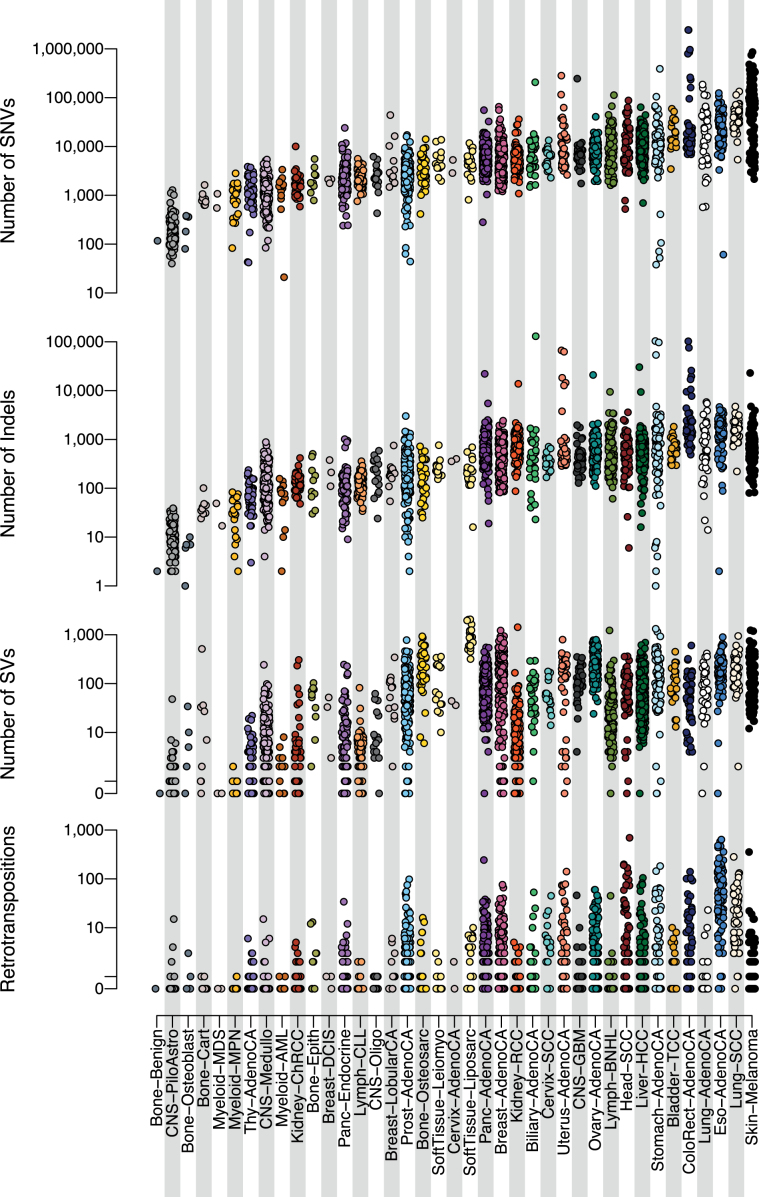
Cancer is driven by genetic change, and the advent of massively parallel sequencing has enabled systematic documentation of this variation at the whole-genome scale1-3. Here we report the integrative analysis of 2,658 whole-cancer genomes and their matching normal tissues across 38 tumour types from the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA). We describe the generation of the PCAWG resource, facilitated by international data sharing using compute clouds. On average, cancer genomes contained 4-5 driver mutations when combining coding and non-coding genomic elements; however, in around 5% of cases no drivers were identified, suggesting that cancer driver discovery is not yet complete. Chromothripsis, in which many clustered structural variants arise in a single catastrophic event, is frequently an early event in tumour evolution; in acral melanoma, for example, these events precede most somatic point mutations and affect several cancer-associated genes simultaneously. Cancers with abnormal telomere maintenance often originate from tissues with low replicative activity and show several mechanisms of preventing telomere attrition to critical levels. Common and rare germline variants affect patterns of somatic mutation, including point mutations, structural variants and somatic retrotransposition. A collection of papers from the PCAWG Consortium describes non-coding mutations that drive cancer beyond those in the TERT promoter4; identifies new signatures of mutational processes that cause base substitutions, small insertions and deletions and structural variation5,6; analyses timings and patterns of tumour evolution7; describes the diverse transcriptional consequences of somatic mutation on splicing, expression levels, fusion genes and promoter activity8,9; and evaluates a range of more-specialized features of cancer genomes8,10-18.
Conflict of interest statement
Gad Getz receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, MSMutSig and POLYSOLVER. Hikmat Al-Ahmadie is consultant for AstraZeneca and Bristol-Myers Squibb. Samuel Aparicio is a founder and shareholder of Contextual Genomics. Pratiti Bandopadhayay receives grant funding from Novartis for an unrelated project. Rameen Beroukhim owns equity in Ampressa Therapeutics. Andrew Biankin receives grant funding from Celgene, AstraZeneca and is a consultant for or on advisory boards of AstraZeneca, Celgene, Elstar Therapeutics, Clovis Oncology and Roche. Ewan Birney is a consultant for Oxford Nanopore, Dovetail and GSK. Marcus Bosenberg is a consultant for Eli Lilly. Atul Butte is a cofounder of and consultant for Personalis, NuMedii, a consultant for Samsung, Geisinger Health, Mango Tree Corporation, Regenstrief Institute and in the recent past a consultant for 10x Genomics and Helix, a shareholder in Personalis, a minor shareholder in Apple, Twitter, Facebook, Google, Microsoft, Sarepta, 10x Genomics, Amazon, Biogen, CVS, Illumina, Snap and Sutro and has received honoraria and travel reimbursement for invited talks from Genentech, Roche, Pfizer, Optum, AbbVie and many academic institutions and health systems. Carlos Caldas has served on the Scientific Advisory Board of Illumina. Lorraine Chantrill acted on an advisory board for AMGEN Australia in the past 2 years. Andrew D. Cherniack receives research funding from Bayer. Helen Davies is an inventor on a number of patent applications that encompass the use of mutational signatures. Francisco De La Vega was employed at Annai Systems during part of the project. Ronny Drapkin serves on the scientific advisory board of Repare Therapeutics and Siamab Therapeutics. Rosalind Eeles has received an honorarium for the GU-ASCO meeting in San Francisco in January 2016 as a speaker, a honorarium and support from Janssen for the RMH FR meeting in November 2017 as a speaker (title: genetics and prostate cancer), a honorarium for an University of Chicago invited talk in May 2018 as speaker and an educational honorarium paid by Bayer & Ipsen to attend GU Connect ‘Treatment sequencing for mCRPC patients within the changing landscape of mHSPC’ at a venue at ESMO, Barcelona, on 28 September 2019. Paul Flicek is a member of the scientific advisory boards of Fabric Genomics and Eagle Genomics. Ronald Ghossein is a consultant for Veracyte. Dominik Glodzik is an inventor on a number of patent applications that encompass the use of mutational signatures. Eoghan Harrington is a full-time employee of Oxford Nanopore Technologies and is a stock holder. Yann Joly is responsible for the Data Access Compliance Office (DACO) of ICGC 2009-2018. Sissel Juul is a full-time employee of Oxford Nanopore Technologies and is a stock holder. Vincent Khoo has received personal fees and non-financial support from Accuray, Astellas, Bayer, Boston Scientific and Janssen. Stian Knappskog is a coprincipal investigator on a clinical trial that receives research funding from AstraZeneca and Pfizer. Ignaty Leshchiner is a consultant for PACT Pharma. Carlos López-Otín has ownership interest (including stock and patents) in DREAMgenics. Matthew Meyerson is a scientific advisory board chair of, and consultant for, OrigiMed, has obtained research funding from Bayer and Ono Pharma and receives patent royalties from LabCorp. Serena Nik-Zainal is an inventor on a number of patent applications that encompass the use of mutational signatures. Nathan Pennell has done consulting work with Merck, Astrazeneca, Eli Lilly and Bristol-Myers Squibb. Xose S. Puente has ownership interest (including stock and patents in DREAMgenics. Benjamin J. Raphael is a consultant for and has ownership interest (including stock and patents) in Medley Genomics. Jorge Reis-Filho is a consultant for Goldman Sachs and REPARE Therapeutics, member of the scientific advisory board of Volition RX and Paige.AI and an ad hoc member of the scientific advisory board of Ventana Medical Systems, Roche Tissue Diagnostics, InVicro, Roche, Genentech and Novartis. Lewis R. Roberts has received grant support from ARIAD Pharmaceuticals, Bayer, BTG International, Exact Sciences, Gilead Sciences, Glycotest, RedHill Biopharma, Target PharmaSolutions and Wako Diagnostics and has provided advisory services to Bayer, Exact Sciences, Gilead Sciences, GRAIL, QED Therapeutics and TAVEC Pharmaceuticals. Richard A. Scolyer has received fees for professional services from Merck Sharp & Dohme, GlaxoSmithKline Australia, Bristol-Myers Squibb, Dermpedia, Novartis Pharmaceuticals Australia, Myriad, NeraCare GmbH and Amgen. Tal Shmaya is employed at Annai Systems. Reiner Siebert has received speaker honoraria from Roche and AstraZeneca. Sabina Signoretti is a consultant for Bristol-Myers Squibb, AstraZeneca, Merck, AACR and NCI and has received funding from Bristol-Myers Squibb, AstraZeneca, Exelixis and royalties from Biogenex. Jared Simpson has received research funding and travel support from Oxford Nanopore Technologies. Anil K. Sood is a consultant for Merck and Kiyatec, has received research funding from M-Trap and is a shareholder in BioPath. Simon Tavaré is on the scientific advisory board of Ipsen and a consultant for Kallyope. John F. Thompson has received honoraria and travel support for attending advisory board meetings of GlaxoSmithKline and Provectus and has received honoraria for participation in advisory boards for MSD Australia and BMS Australia. Daniel Turner is a full-time employee of Oxford Nanopore Technologies and is a stock holder. Naveen Vasudev has received speaker honoraria and/or consultancy fees from Bristol-Myers Squibb, Pfizer, EUSA pharma, MSD and Novartis. Jeremiah A. Wala is a consultant for Nference. Daniel J. Weisenberger is a consultant for Zymo Research. Dai-Ying Wu is employed at Annai Systems. Cheng-Zhong Zhang is a cofounder and equity holder of Pillar Biosciences, a for-profit company that specializes in the development of targeted sequencing assays. The other authors declare no competing interests.
Figures
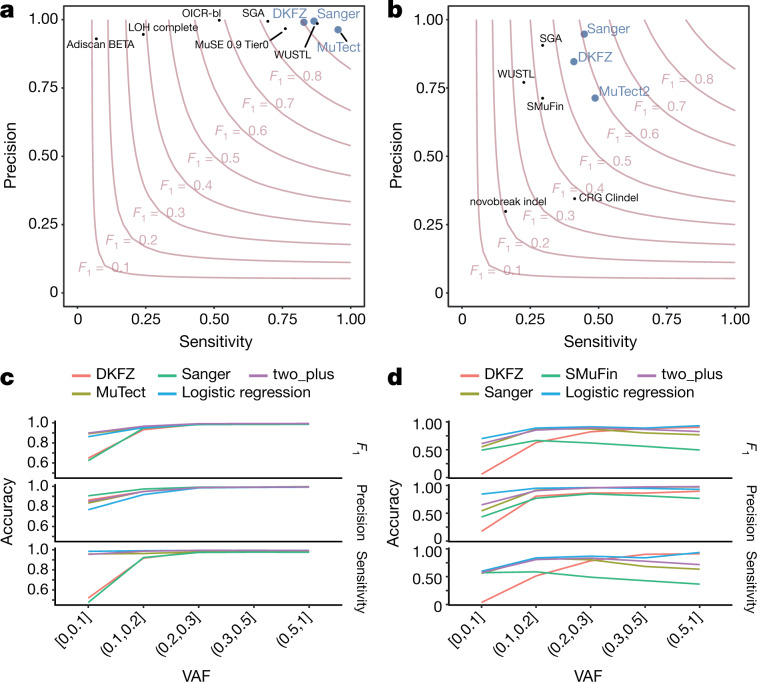
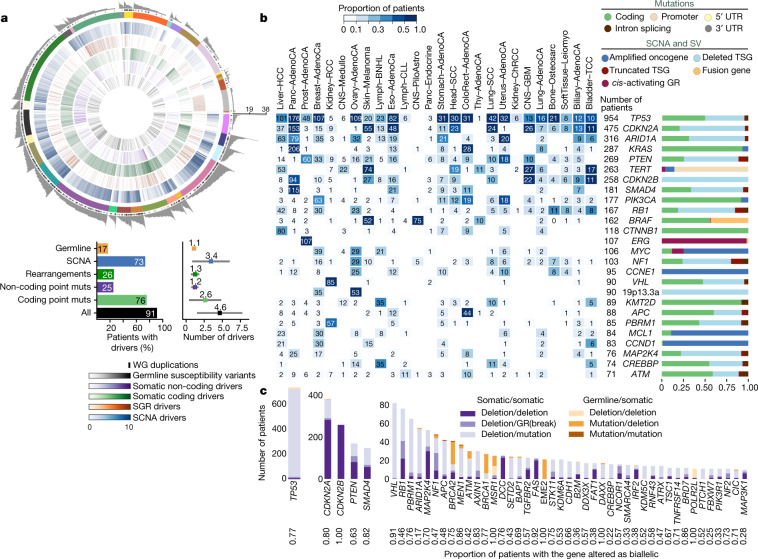
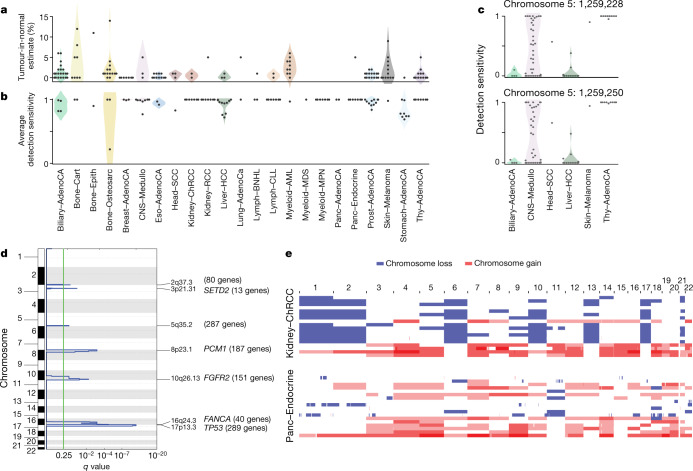
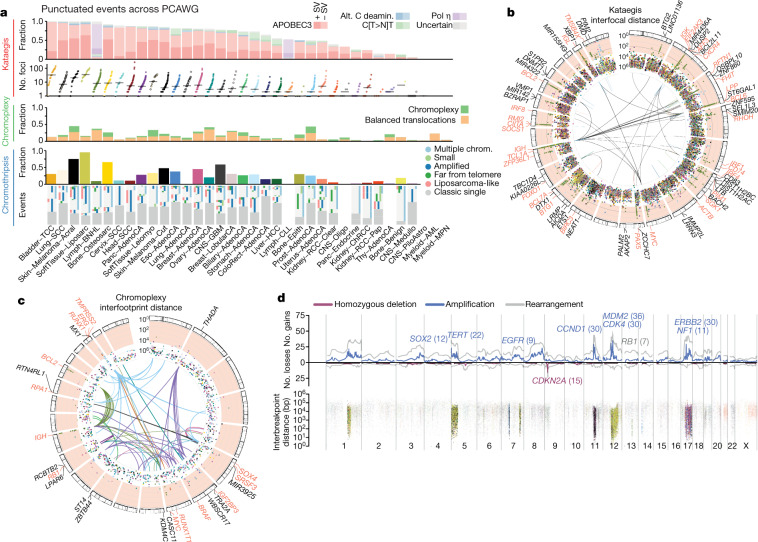


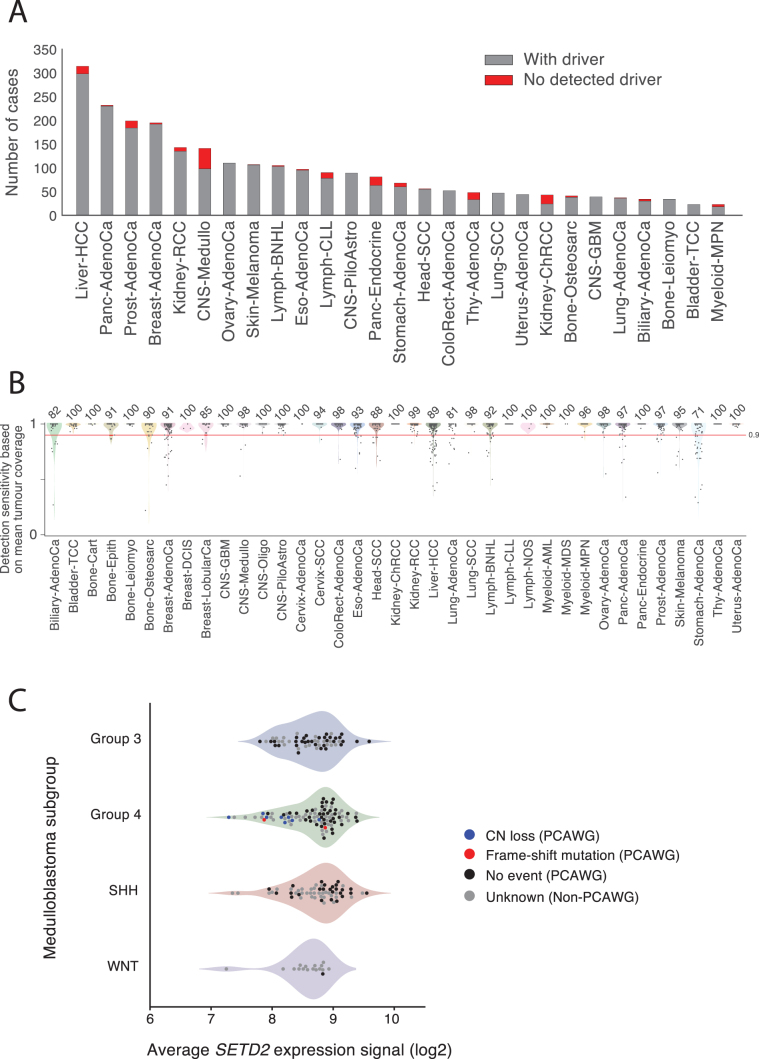
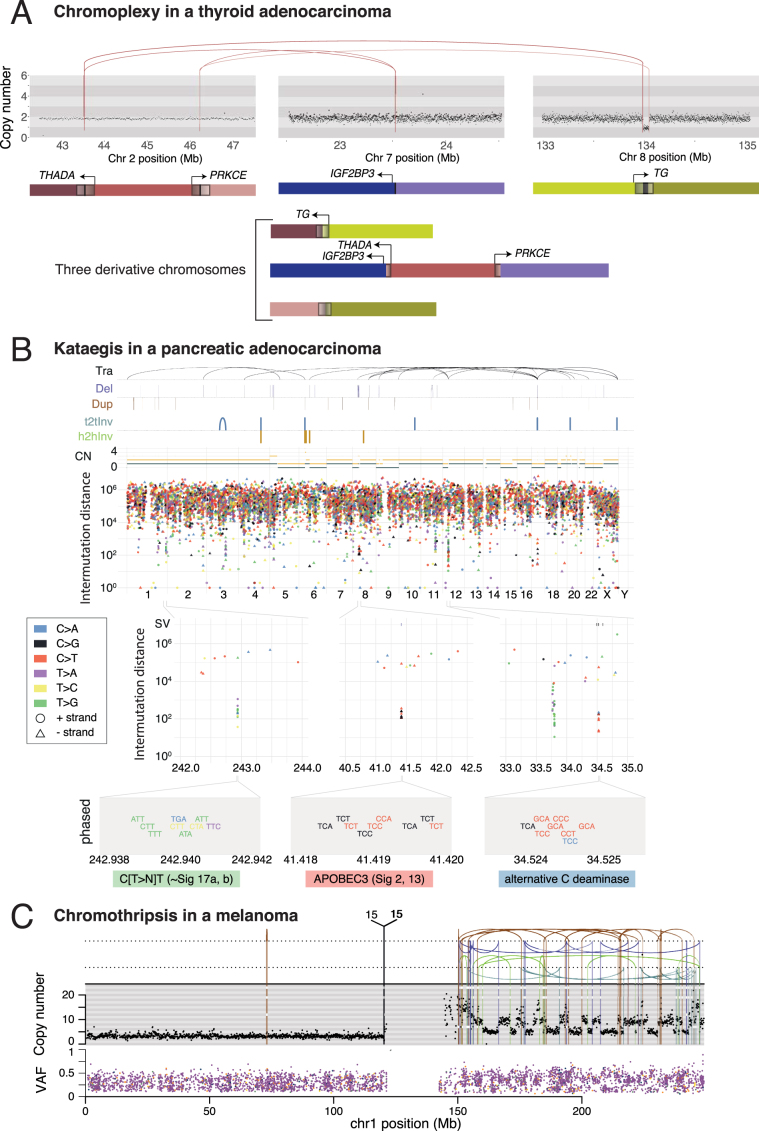
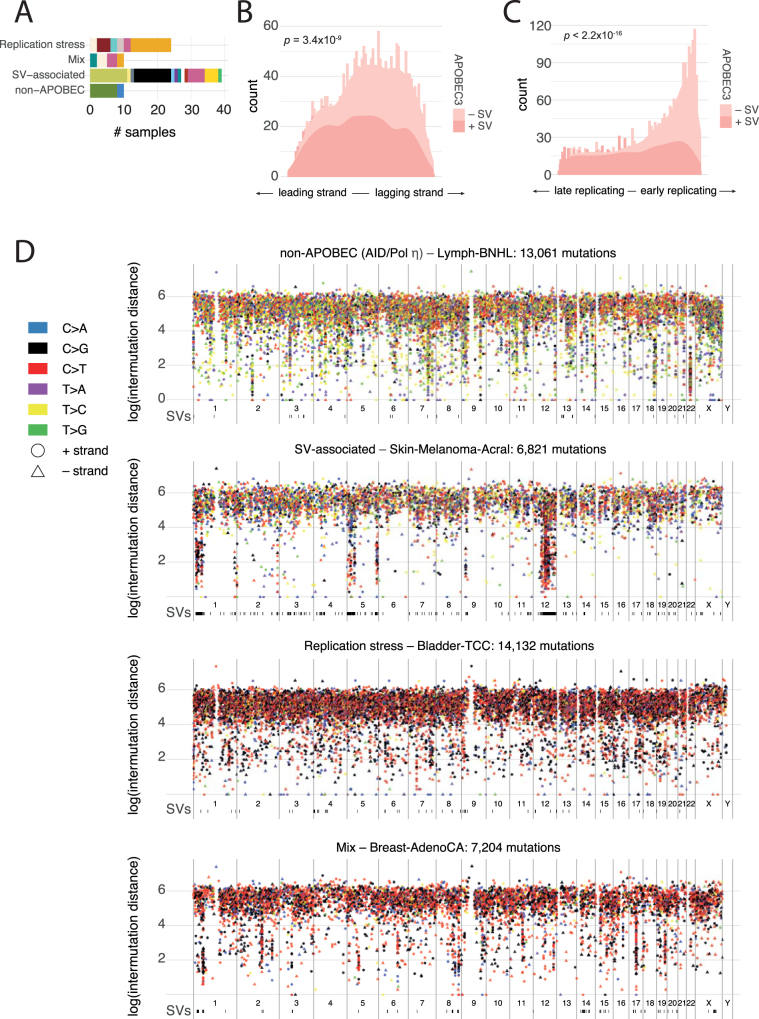

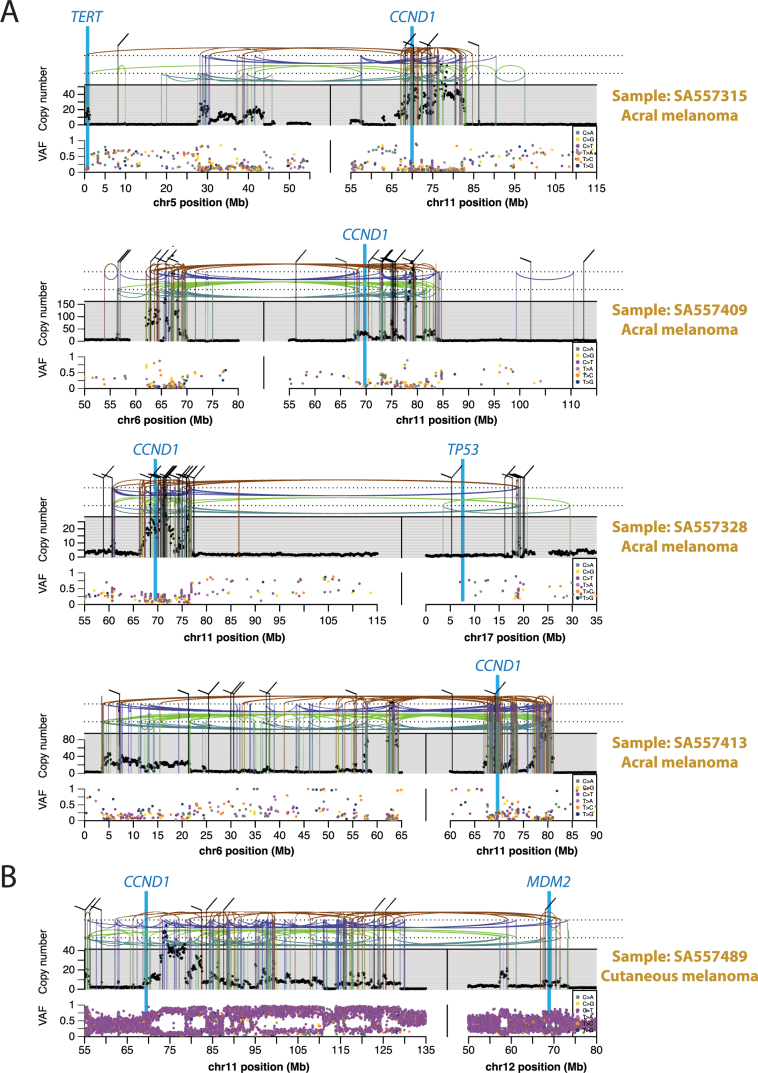
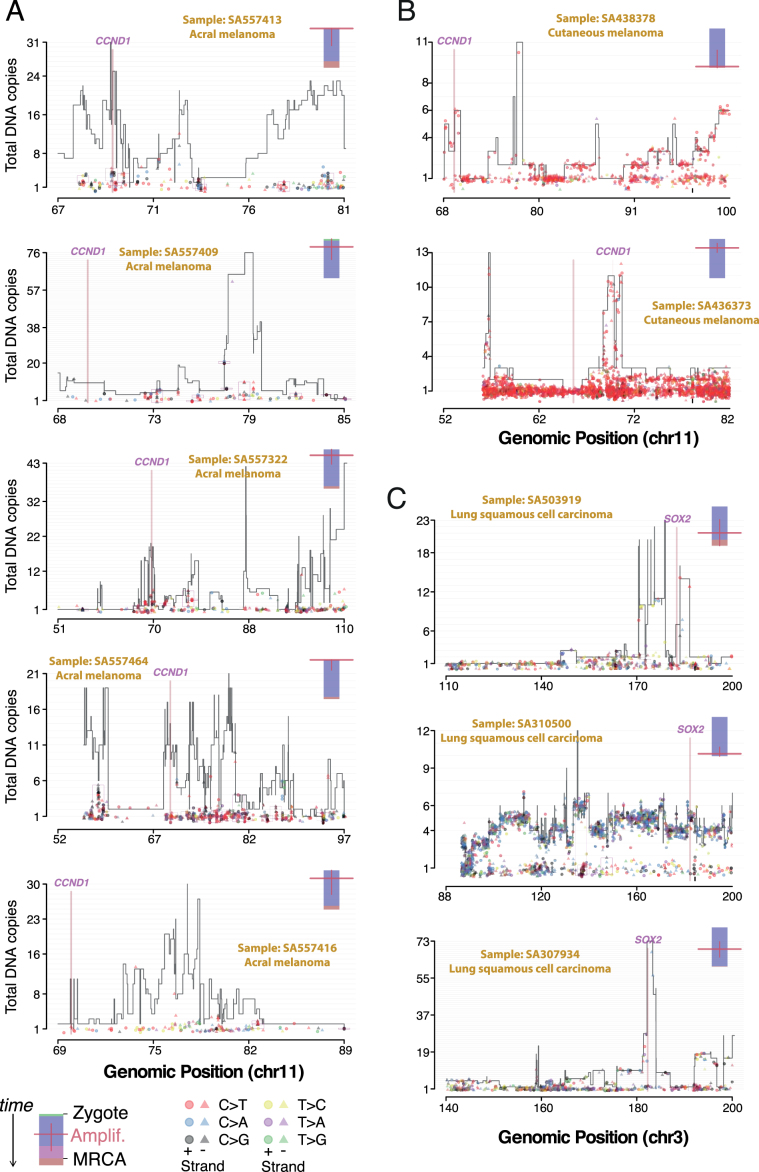

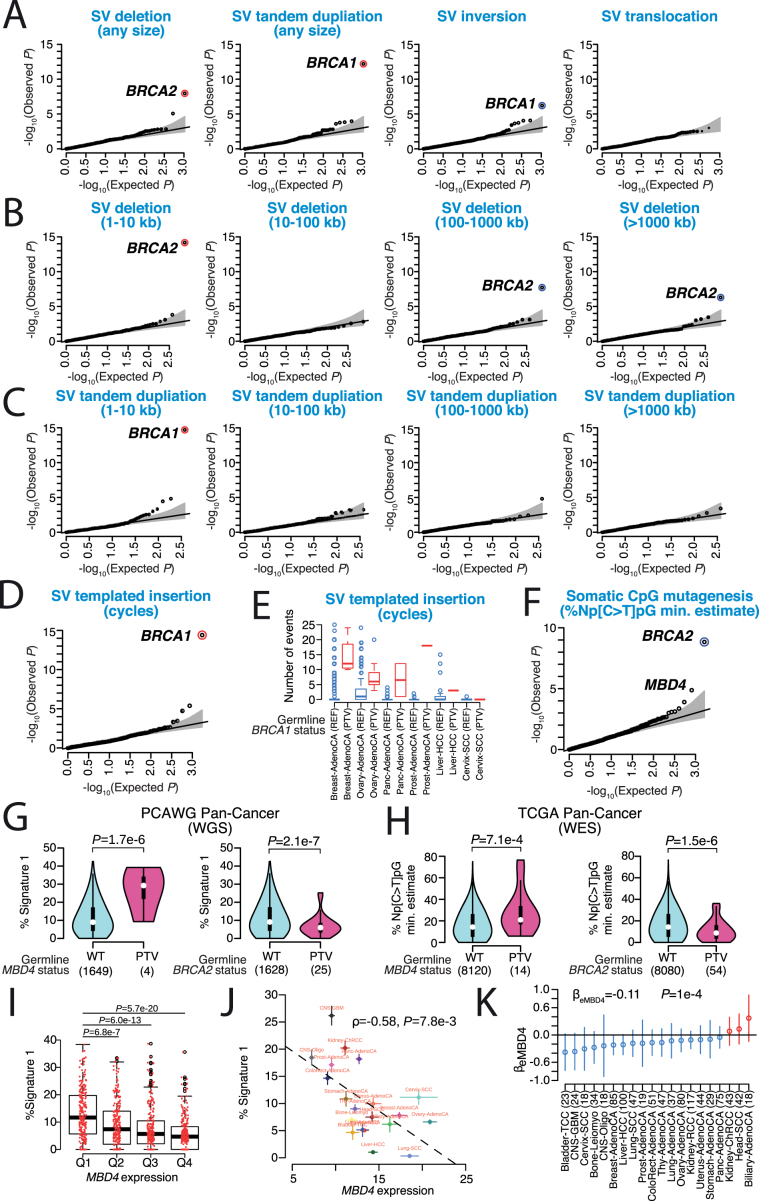


Comment in
-
Global genomics project unravels cancer's complexity at unprecedented scale.Nature. 2020 Feb;578(7793):39-40. doi: 10.1038/d41586-020-00213-2. Nature. 2020. PMID: 32025004 No abstract available.
Similar articles
-
Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing.Nat Genet. 2020 Mar;52(3):331-341. doi: 10.1038/s41588-019-0576-7. Epub 2020 Feb 5. Nat Genet. 2020. PMID: 32025003 Free PMC article.
-
Patterns of somatic structural variation in human cancer genomes.Nature. 2020 Feb;578(7793):112-121. doi: 10.1038/s41586-019-1913-9. Epub 2020 Feb 5. Nature. 2020. PMID: 32025012 Free PMC article.
-
Analyses of non-coding somatic drivers in 2,658 cancer whole genomes.Nature. 2020 Feb;578(7793):102-111. doi: 10.1038/s41586-020-1965-x. Epub 2020 Feb 5. Nature. 2020. PMID: 32025015 Free PMC article.
-
Decoding human cancer with whole genome sequencing: a review of PCAWG Project studies published in February 2020.Cancer Metastasis Rev. 2021 Sep;40(3):909-924. doi: 10.1007/s10555-021-09969-z. Epub 2021 Jun 7. Cancer Metastasis Rev. 2021. PMID: 34097189 Free PMC article. Review.
-
Beyond the exome: the role of non-coding somatic mutations in cancer.Ann Oncol. 2016 Feb;27(2):240-8. doi: 10.1093/annonc/mdv561. Epub 2015 Nov 23. Ann Oncol. 2016. PMID: 26598542 Review.
Cited by
-
Tumor organoids improve mutation detection of pancreatic ductal adenocarcinoma.Sci Rep. 2024 Oct 26;14(1):25468. doi: 10.1038/s41598-024-75888-y. Sci Rep. 2024. PMID: 39462012 Free PMC article.
-
The prominent pervasive oncogenic role and tissue specific permissiveness of RAS gene mutations.Sci Rep. 2024 Oct 26;14(1):25452. doi: 10.1038/s41598-024-76591-8. Sci Rep. 2024. PMID: 39455841 Free PMC article.
-
MoNETA: MultiOmics Network Embedding for SubType Analysis.NAR Genom Bioinform. 2024 Oct 16;6(4):lqae141. doi: 10.1093/nargab/lqae141. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39416887 Free PMC article.
-
The baseline hemoglobin level is a positive biomarker for immunotherapy response and can improve the predictability of tumor mutation burden for immunotherapy response in cancer.Front Pharmacol. 2024 Oct 2;15:1456833. doi: 10.3389/fphar.2024.1456833. eCollection 2024. Front Pharmacol. 2024. PMID: 39415833 Free PMC article.
-
Comprehensive evaluation and guidance of structural variation detection tools in chicken whole genome sequence data.BMC Genomics. 2024 Oct 16;25(1):970. doi: 10.1186/s12864-024-10875-1. BMC Genomics. 2024. PMID: 39415108 Free PMC article.
References
MeSH terms
Substances
Grants and funding
- 15874/CRUK_/Cancer Research UK/United Kingdom
- 27815/CRUK_/Cancer Research UK/United Kingdom
- U24 CA210950/CA/NCI NIH HHS/United States
- 18387/CRUK_/Cancer Research UK/United Kingdom
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- 22720/CRUK_/Cancer Research UK/United Kingdom
- T32 GM008313/GM/NIGMS NIH HHS/United States
- R35 GM127029/GM/NIGMS NIH HHS/United States
- 23433/CRUK_/Cancer Research UK/United Kingdom
- U24 CA211000/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- 20952/CRUK_/Cancer Research UK/United Kingdom
- 23916/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00016/11/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/16/MRC_/Medical Research Council/United Kingdom
- R01 CA235162/CA/NCI NIH HHS/United States
- G0300648/MRC_/Medical Research Council/United Kingdom
- 14545/CRUK_/Cancer Research UK/United Kingdom
- 16942/CRUK_/Cancer Research UK/United Kingdom
- 27176/CRUK_/Cancer Research UK/United Kingdom
- MC_U137961146/MRC_/Medical Research Council/United Kingdom
- 22932/CRUK_/Cancer Research UK/United Kingdom
- R01 GM109031/GM/NIGMS NIH HHS/United States
- R01 HG007069/HG/NHGRI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- U01 CA217842/CA/NCI NIH HHS/United States
- RIF2015_A06_JAMIESON/PANCREATICCANUK_/Pancreatic Cancer UK/United Kingdom
- U24 CA180951/CA/NCI NIH HHS/United States
- R01 CA218668/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- 22131/CRUK_/Cancer Research UK/United Kingdom
- G1000729/MRC_/Medical Research Council/United Kingdom
- 23924/CRUK_/Cancer Research UK/United Kingdom
- P01 CA240239/CA/NCI NIH HHS/United States
- R01 CA183793/CA/NCI NIH HHS/United States
- P30 CA014236/CA/NCI NIH HHS/United States
- MR/L008963/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12022/2/MRC_/Medical Research Council/United Kingdom
- U24 CA210949/CA/NCI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
- 23917/CRUK_/Cancer Research UK/United Kingdom
- 25813/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_12009/11/MRC_/Medical Research Council/United Kingdom
- 26718/CRUK_/Cancer Research UK/United Kingdom
- UG1 CA233339/CA/NCI NIH HHS/United States
- 088177/WT_/Wellcome Trust/United Kingdom
- U24 CA210990/CA/NCI NIH HHS/United States
