

CSB promoter downregulation via histone H3 hypoacetylation is an early determinant of replicative senescence
- PMID: 31811121
- PMCID: PMC6898346
- DOI: 10.1038/s41467-019-13314-y
CSB promoter downregulation via histone H3 hypoacetylation is an early determinant of replicative senescence
Abstract
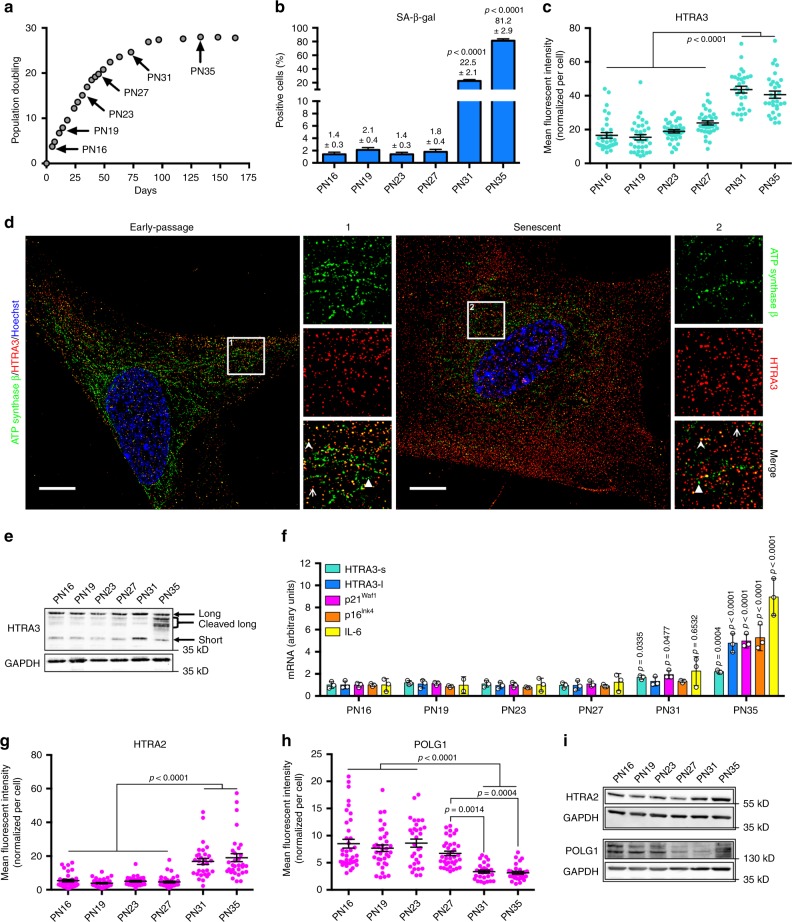
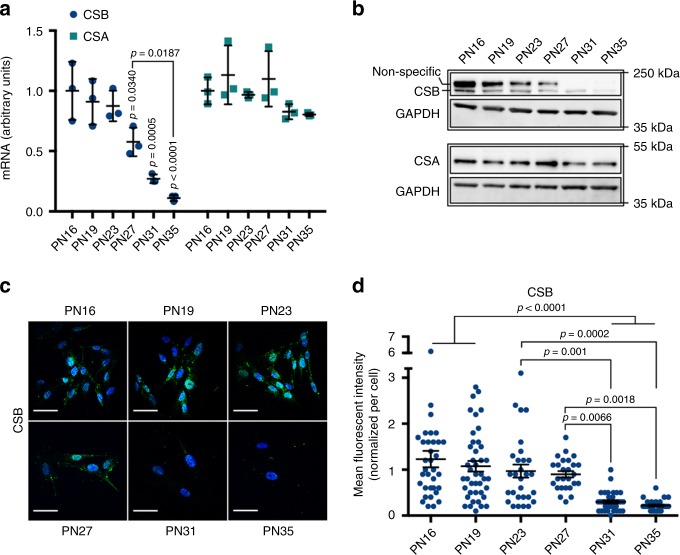
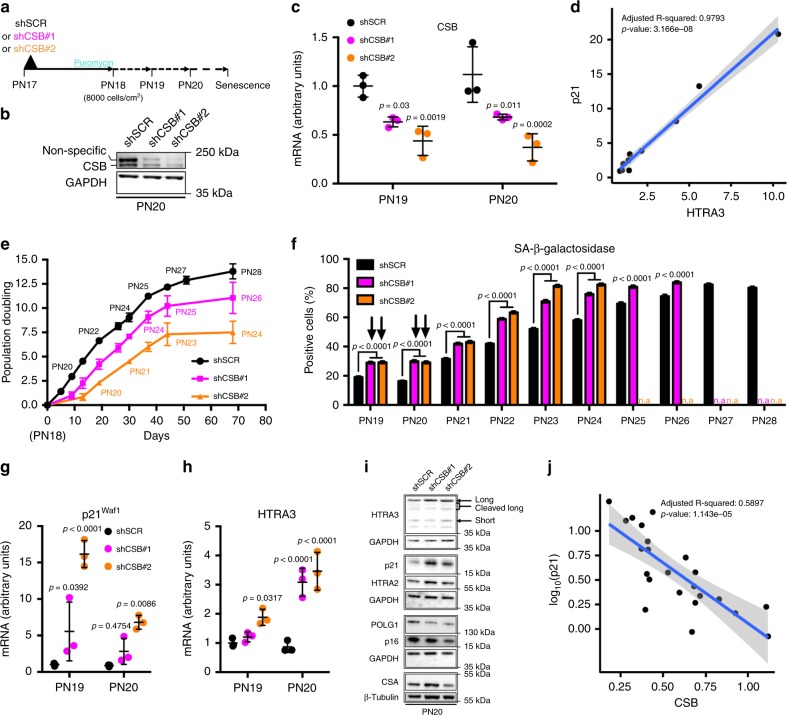
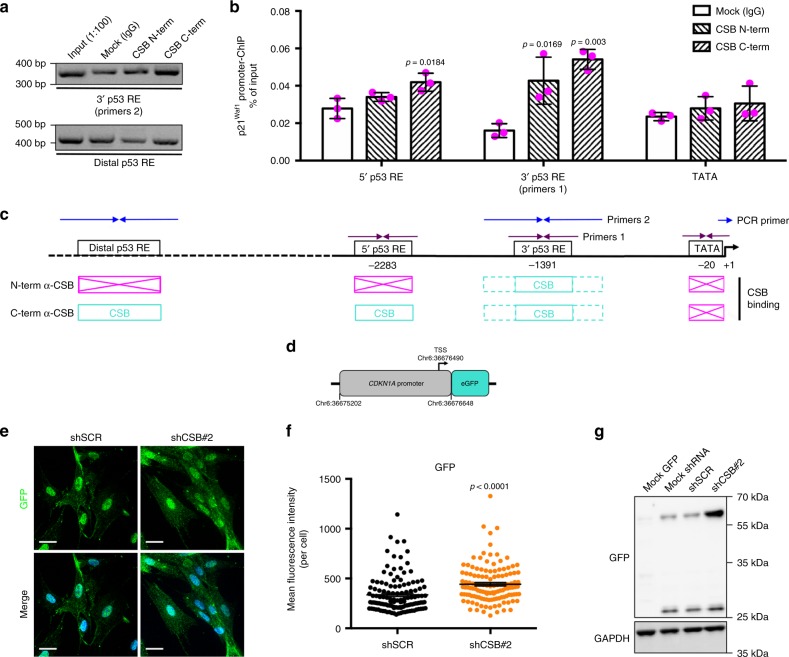
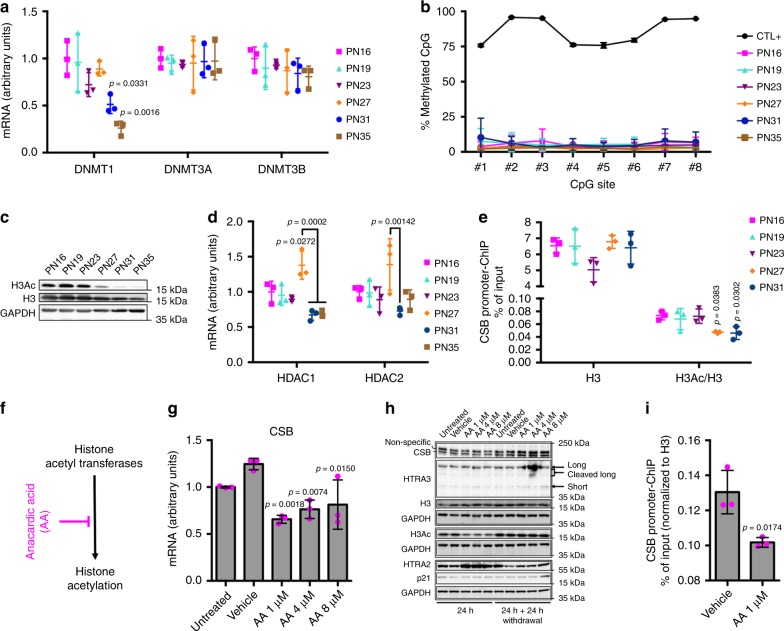
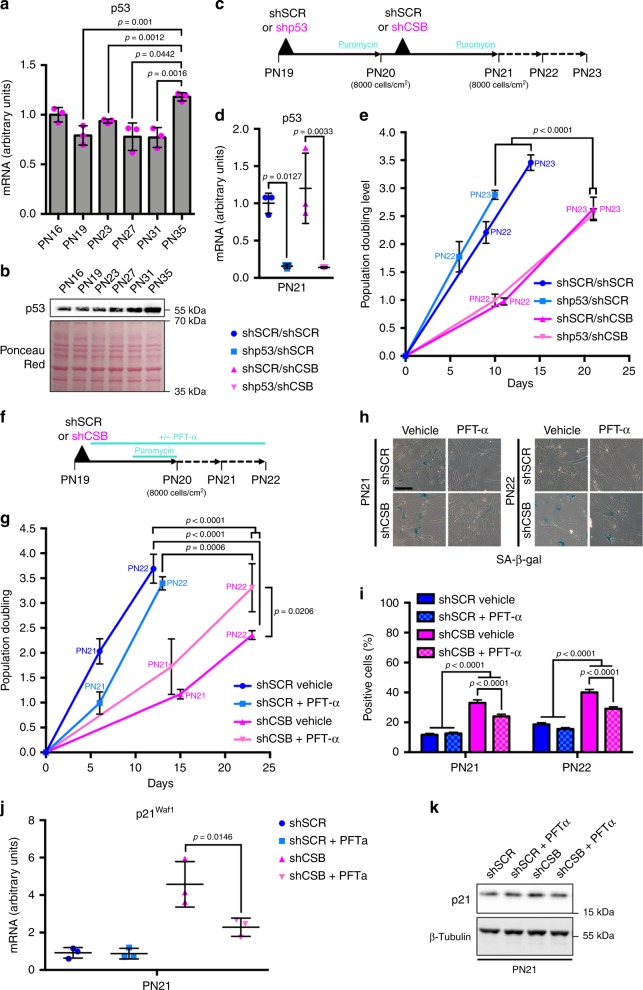
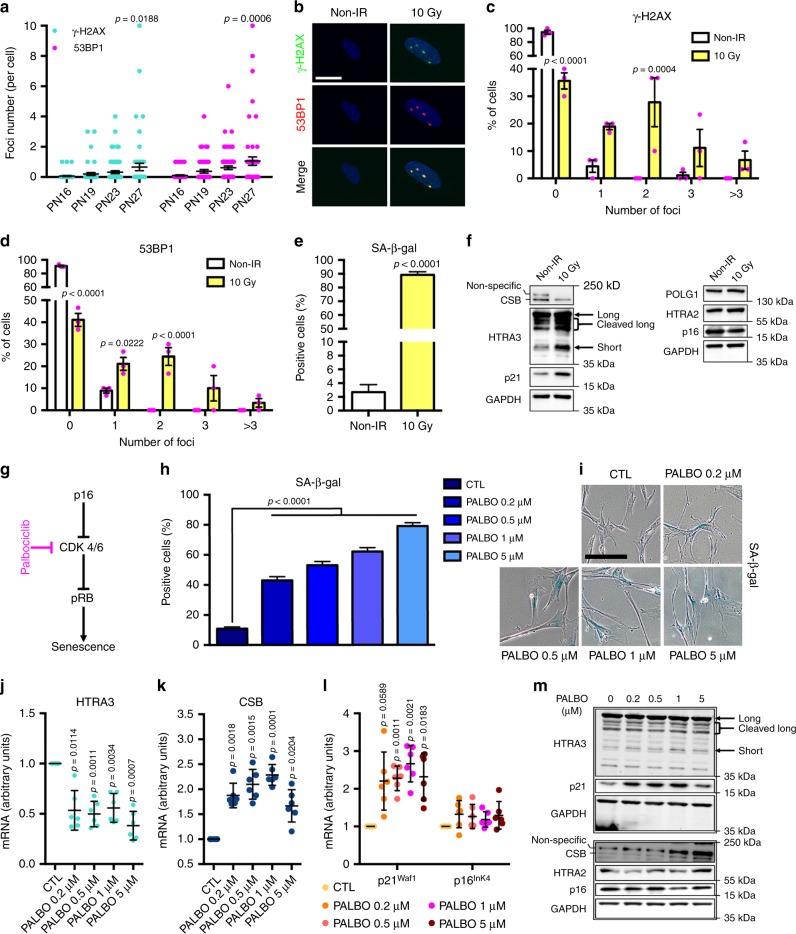
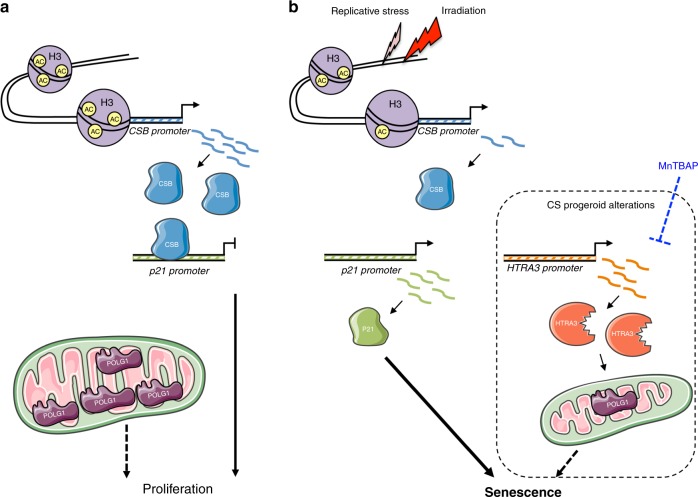
Cellular senescence has causative links with ageing and age-related diseases, however, it remains unclear if progeroid factors cause senescence in normal cells. Here, we show that depletion of CSB, a protein mutated in progeroid Cockayne syndrome (CS), is the earliest known trigger of p21-dependent replicative senescence. CSB depletion promotes overexpression of the HTRA3 protease resulting in mitochondrial impairments, which are causally linked to CS pathological phenotypes. The CSB promoter is downregulated by histone H3 hypoacetylation during DNA damage-response. Mechanistically, CSB binds to the p21 promoter thereby downregulating its transcription and blocking replicative senescence in a p53-independent manner. This activity of CSB is independent of its role in the repair of UV-induced DNA damage. HTRA3 accumulation and senescence are partially rescued upon reduction of oxidative/nitrosative stress. These findings establish a CSB/p21 axis that acts as a barrier to replicative senescence, and link a progeroid factor with the process of regular ageing in human.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging.Nucleic Acids Res. 2019 Sep 19;47(16):8548-8562. doi: 10.1093/nar/gkz568. Nucleic Acids Res. 2019. PMID: 31276581 Free PMC article.
-
Poly(ADP-ribose) polymerase 1 (PARP1) promotes oxidative stress-induced association of Cockayne syndrome group B protein with chromatin.J Biol Chem. 2018 Nov 16;293(46):17863-17874. doi: 10.1074/jbc.RA118.004548. Epub 2018 Sep 28. J Biol Chem. 2018. PMID: 30266807 Free PMC article.
-
Cockayne syndrome group A and ferrochelatase finely tune ribosomal gene transcription and its response to UV irradiation.Nucleic Acids Res. 2021 Nov 8;49(19):10911-10930. doi: 10.1093/nar/gkab819. Nucleic Acids Res. 2021. PMID: 34581821 Free PMC article.
-
Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer.Cells. 2021 Apr 10;10(4):866. doi: 10.3390/cells10040866. Cells. 2021. PMID: 33920220 Free PMC article. Review.
-
The role of CSA and CSB protein in the oxidative stress response.Mech Ageing Dev. 2013 May-Jun;134(5-6):261-9. doi: 10.1016/j.mad.2013.03.006. Epub 2013 Apr 3. Mech Ageing Dev. 2013. PMID: 23562424 Review.
Cited by
-
Beyond DNA repair and chromosome instability-Fanconi anaemia as a cellular senescence-associated syndrome.Cell Death Differ. 2021 Apr;28(4):1159-1173. doi: 10.1038/s41418-021-00764-5. Epub 2021 Mar 15. Cell Death Differ. 2021. PMID: 33723374 Free PMC article. Review.
-
Effects of Oxygen Tension for Membrane Lipidome Remodeling of Cockayne Syndrome Cell Models.Cells. 2022 Apr 10;11(8):1286. doi: 10.3390/cells11081286. Cells. 2022. PMID: 35455966 Free PMC article.
-
Multiple functions of p21 in cancer radiotherapy.J Cancer Res Clin Oncol. 2021 Apr;147(4):987-1006. doi: 10.1007/s00432-021-03529-2. Epub 2021 Feb 5. J Cancer Res Clin Oncol. 2021. PMID: 33547489 Review.
-
DNA-PKcs regulates myogenesis in an Akt-dependent manner independent of induced DNA damage.Cell Death Differ. 2023 Aug;30(8):1900-1915. doi: 10.1038/s41418-023-01177-2. Epub 2023 Jul 3. Cell Death Differ. 2023. PMID: 37400716 Free PMC article.
-
Premature aging in genetic diseases: what conclusions can be drawn for physiological aging.Front Aging. 2024 Feb 28;4:1327833. doi: 10.3389/fragi.2023.1327833. eCollection 2023. Front Aging. 2024. PMID: 38481648 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
