

Emerging consensus on the collapse of unfolded and intrinsically disordered proteins in water
- PMID: 31805437
- PMCID: PMC7472963
- DOI: 10.1016/j.sbi.2019.10.009
Emerging consensus on the collapse of unfolded and intrinsically disordered proteins in water
Abstract
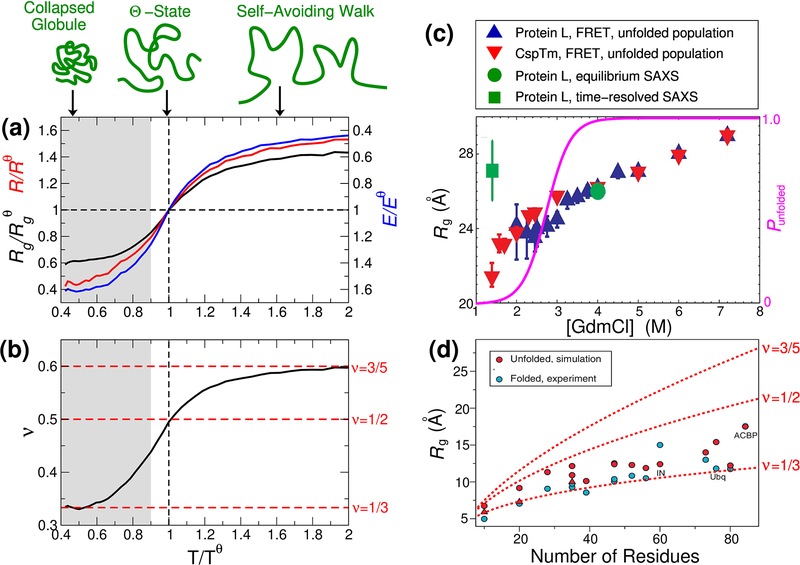
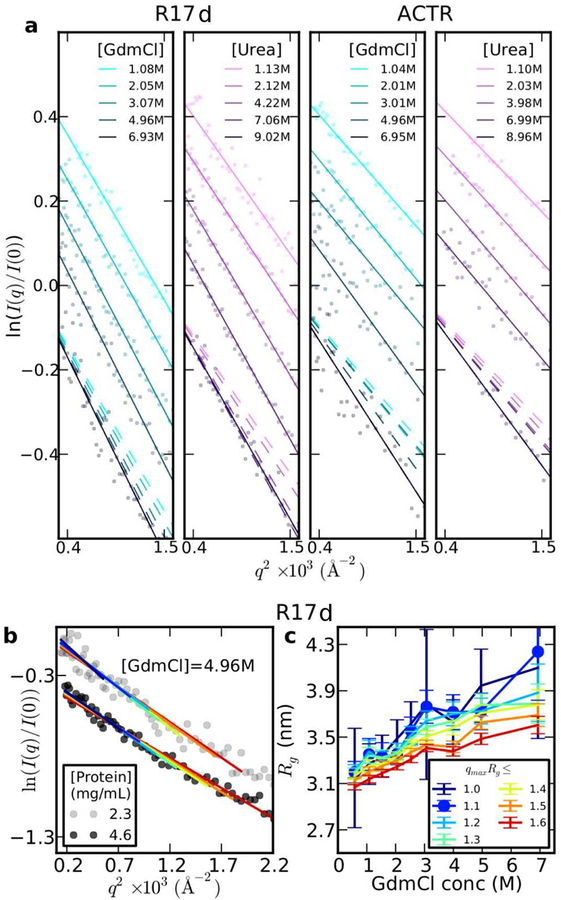
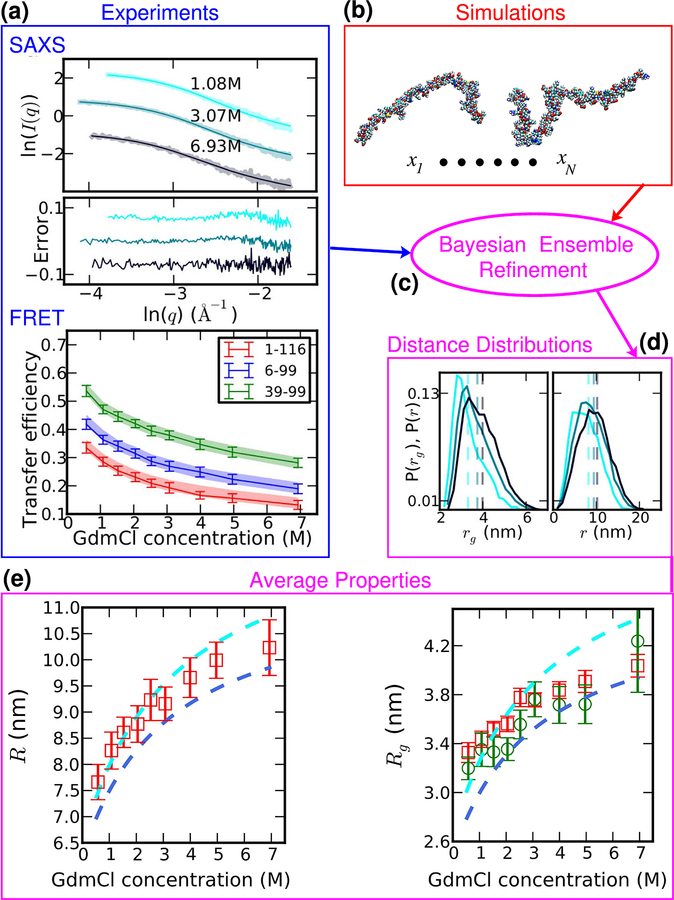
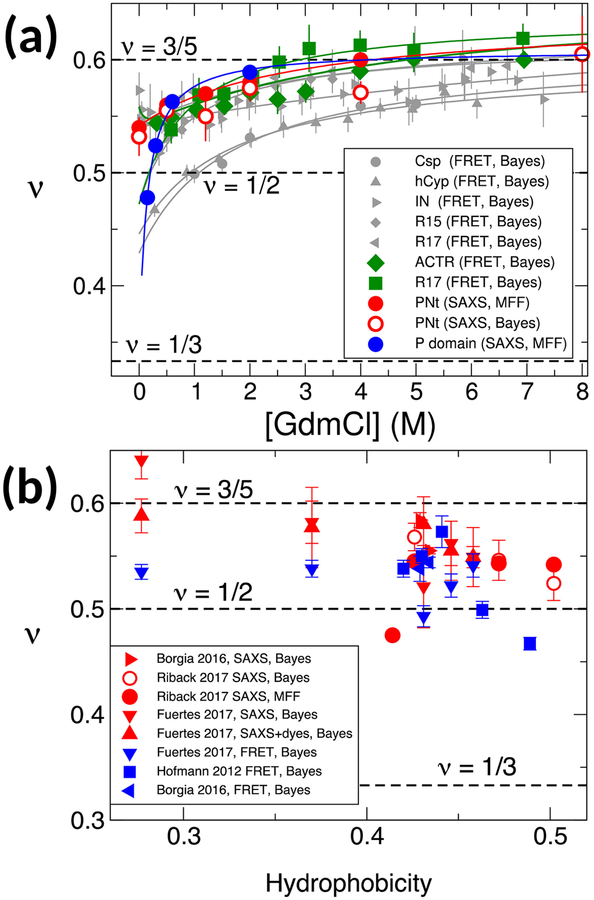
Establishing the degree of collapse of unfolded or disordered proteins is a fundamental problem in biophysics, because of its relation to protein folding and to the function of intrinsically disordered proteins. However, until recently, different experiments gave qualitatively different results on collapse and there were large discrepancies between experiments and all-atom simulations. New methodology introduced in the past three years has helped to resolve the differences between experiments, and improvements in simulations have closed the gap between experiment and simulation. These advances have led to an emerging consensus on the collapse of disordered proteins in water.
Published by Elsevier Ltd.
Conflict of interest statement
Figures
Similar articles
-
Perspective: Chain dynamics of unfolded and intrinsically disordered proteins from nanosecond fluorescence correlation spectroscopy combined with single-molecule FRET.J Chem Phys. 2018 Jul 7;149(1):010901. doi: 10.1063/1.5037683. J Chem Phys. 2018. PMID: 29981536
-
Evolution of All-Atom Protein Force Fields to Improve Local and Global Properties.J Phys Chem Lett. 2019 May 2;10(9):2227-2234. doi: 10.1021/acs.jpclett.9b00850. Epub 2019 Apr 22. J Phys Chem Lett. 2019. PMID: 30990694 Free PMC article.
-
Modest influence of FRET chromophores on the properties of unfolded proteins.Biophys J. 2014 Oct 7;107(7):1654-60. doi: 10.1016/j.bpj.2014.07.071. Biophys J. 2014. PMID: 25296318 Free PMC article.
-
Force field development and simulations of intrinsically disordered proteins.Curr Opin Struct Biol. 2018 Feb;48:40-48. doi: 10.1016/j.sbi.2017.10.008. Epub 2017 Nov 5. Curr Opin Struct Biol. 2018. PMID: 29080468 Free PMC article. Review.
-
Templated folding of intrinsically disordered proteins.J Biol Chem. 2020 May 8;295(19):6586-6593. doi: 10.1074/jbc.REV120.012413. Epub 2020 Apr 6. J Biol Chem. 2020. PMID: 32253236 Free PMC article. Review.
Cited by
-
Full structural ensembles of intrinsically disordered proteins from unbiased molecular dynamics simulations.Commun Biol. 2021 Feb 23;4(1):243. doi: 10.1038/s42003-021-01759-1. Commun Biol. 2021. PMID: 33623120 Free PMC article.
-
Lipid bilayer induces contraction of the denatured state ensemble of a helical-bundle membrane protein.Proc Natl Acad Sci U S A. 2022 Jan 4;119(1):e2109169119. doi: 10.1073/pnas.2109169119. Proc Natl Acad Sci U S A. 2022. PMID: 34969836 Free PMC article.
-
Characterization of Intrinsically Disordered Proteins in Healthy and Diseased States by Nuclear Magnetic Resonance.Rev Recent Clin Trials. 2024;19(3):176-188. doi: 10.2174/0115748871271420240213064251. Rev Recent Clin Trials. 2024. PMID: 38409704 Review.
-
Identification of an α-MoRF in the Intrinsically Disordered Region of the Escargot Transcription Factor.ACS Omega. 2020 Jul 17;5(29):18331-18341. doi: 10.1021/acsomega.0c02051. eCollection 2020 Jul 28. ACS Omega. 2020. PMID: 32743208 Free PMC article.
-
Visualizing and trapping transient oligomers in amyloid assembly pathways.Biophys Chem. 2021 Jan;268:106505. doi: 10.1016/j.bpc.2020.106505. Epub 2020 Nov 10. Biophys Chem. 2021. PMID: 33220582 Free PMC article. Review.
References
-
- Rubinstein M, Colby RH. Polymer Physics. Oxford University Press, Oxford, 2003.
-
- Alonso DOV, Dill KA. Solvent denaturation and stabilization of globular proteins. Biochemistry 1991, 30: 5974–5985. - PubMed
-
- Holehouse AS, Pappu RV. Collapse transitions of proteins and the interplay among backbone, sidechain and solvent interactions. Ann Rev Biophys 2018, 47: 19–39. - PMC - PubMed
-
** Recent review of collapse in unfolded and disordered proteins, broken down into contributions from the backbone and different types of side-chain.
-
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41: 415–427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
