

VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection
- PMID: 31694946
- PMCID: PMC6955268
- DOI: 10.1128/JVI.01454-19
VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection
Abstract
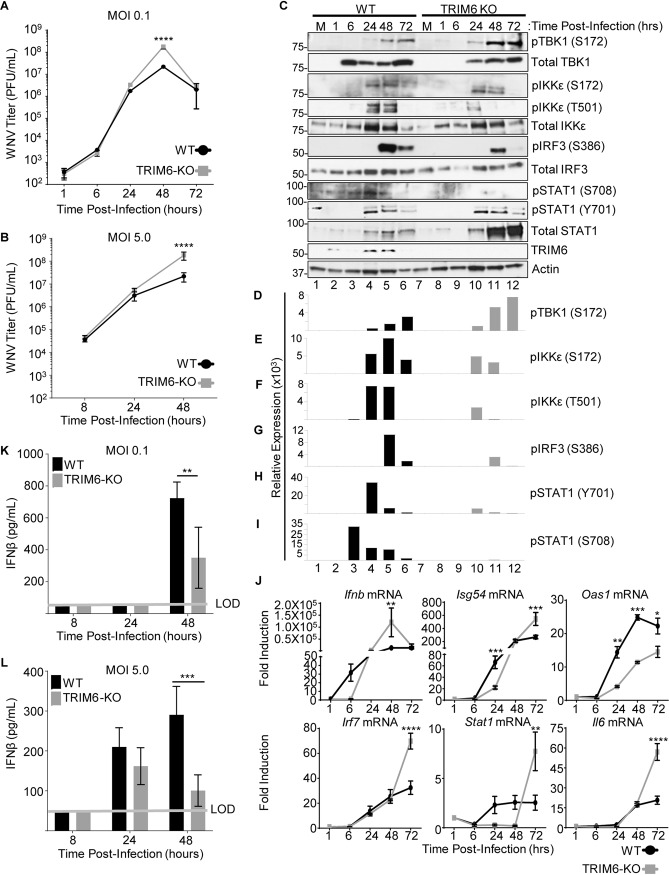
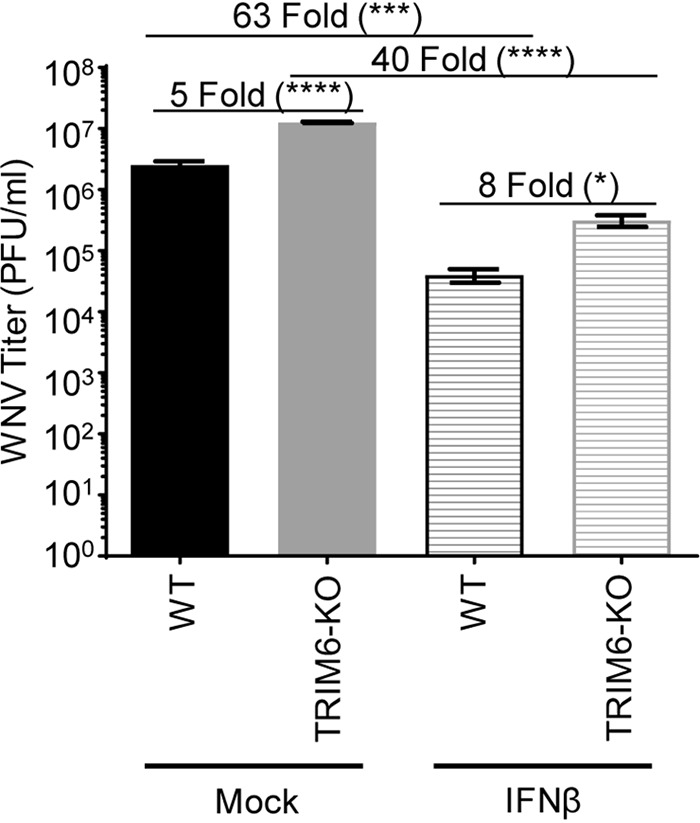
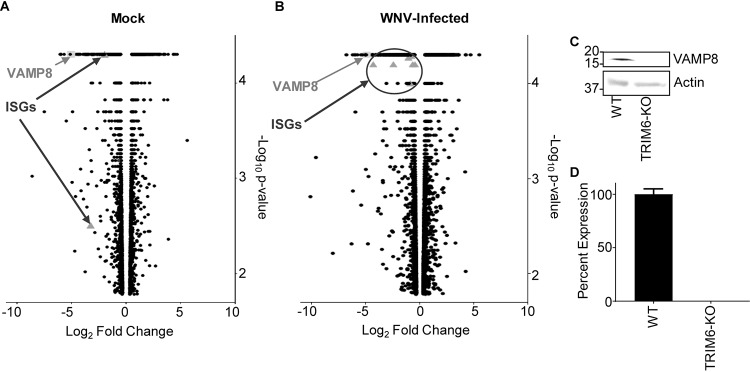
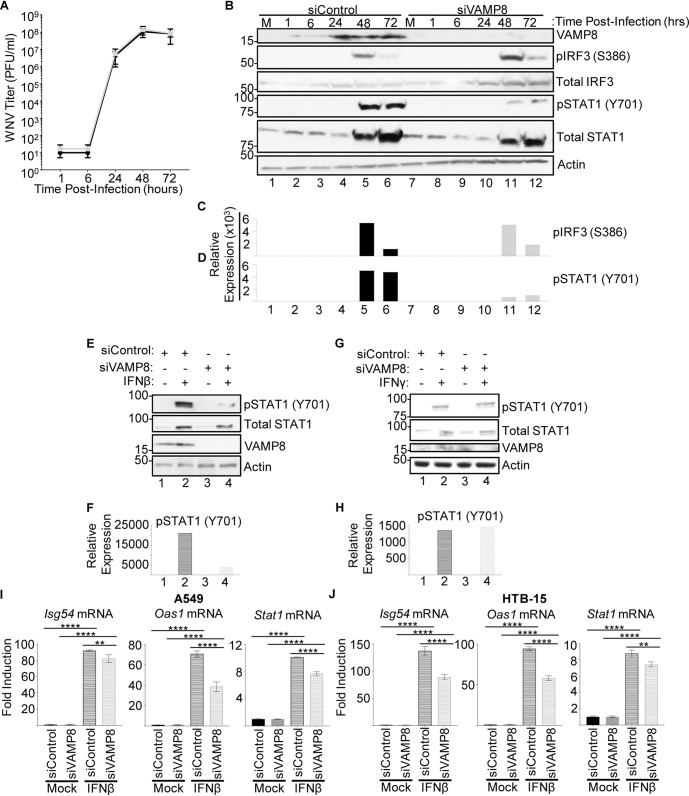
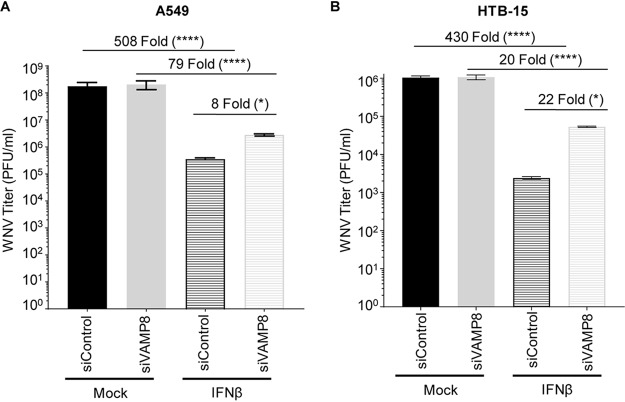
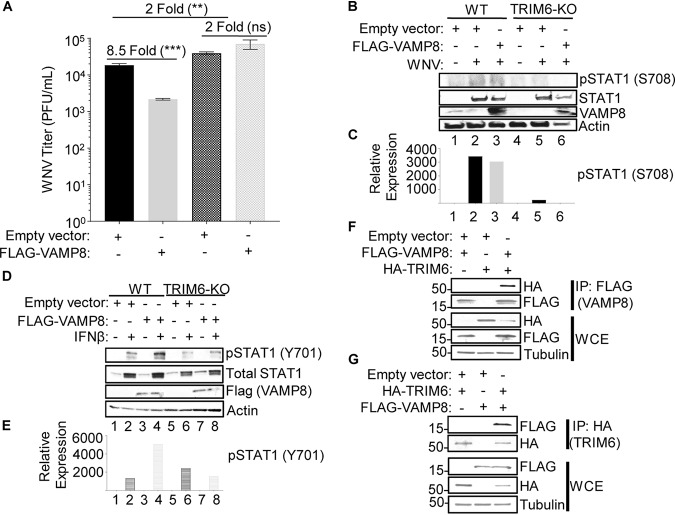
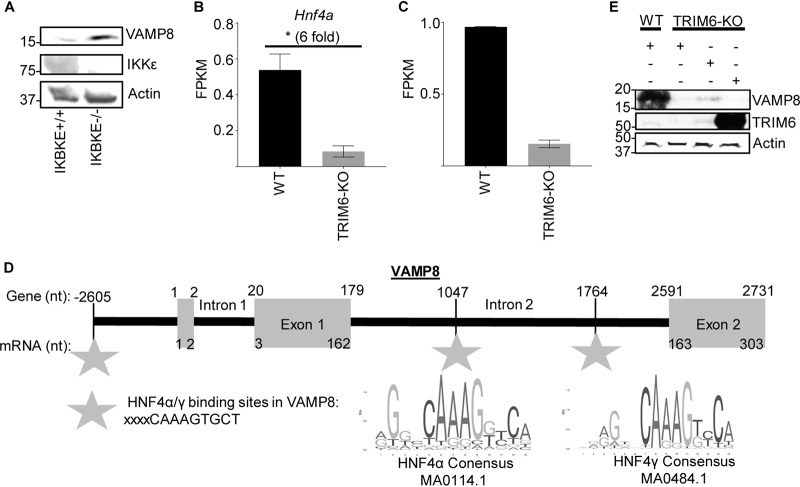
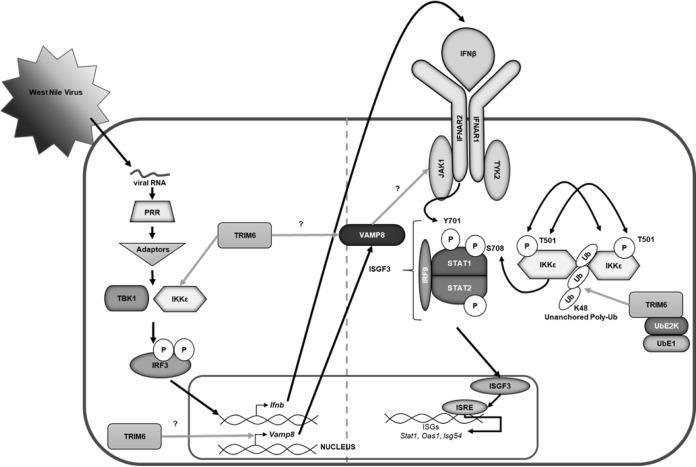
Several members of the tripartite motif (TRIM) family of E3 ubiquitin ligases regulate immune pathways, including the antiviral type I interferon (IFN-I) system. Previously, we demonstrated that TRIM6 is involved in IFN-I induction and signaling. In the absence of TRIM6, optimal IFN-I signaling is reduced, allowing increased replication of interferon-sensitive viruses. Despite having evolved numerous mechanisms to restrict the vertebrate host's IFN-I response, West Nile virus (WNV) replication is sensitive to pretreatment with IFN-I. However, the regulators and products of the IFN-I pathway that are important in regulating WNV replication are incompletely defined. Consistent with WNV's sensitivity to IFN-I, we found that in TRIM6 knockout (TRIM6-KO) A549 cells, WNV replication is significantly increased and IFN-I induction and signaling are impaired compared to wild-type (wt) cells. IFN-β pretreatment was more effective in protecting against subsequent WNV infection in wt cells than TRIM6-KO, indicating that TRIM6 contributes to the establishment of an IFN-induced antiviral response against WNV. Using next-generation sequencing, we identified VAMP8 as a potential factor involved in this TRIM6-mediated antiviral response. VAMP8 knockdown resulted in reduced JAK1 and STAT1 phosphorylation and impaired induction of several interferon-stimulated genes (ISGs) following WNV infection or IFN-β treatment. Furthermore, VAMP8-mediated STAT1 phosphorylation required the presence of TRIM6. Therefore, the VAMP8 protein is a novel regulator of IFN-I signaling, and its expression and function are dependent on TRIM6 activity. Overall, these results provide evidence that TRIM6 contributes to the antiviral response against WNV and identify VAMP8 as a novel regulator of the IFN-I system.IMPORTANCE WNV is a mosquito-borne flavivirus that poses a threat to human health across large discontinuous areas throughout the world. Infection with WNV results in febrile illness, which can progress to severe neurological disease. Currently, there are no approved treatment options to control WNV infection. Understanding the cellular immune responses that regulate viral replication is important in diversifying the resources available to control WNV. Here, we show that the elimination of TRIM6 in human cells results in an increase in WNV replication and alters the expression and function of other components of the IFN-I pathway through VAMP8. Dissecting the interactions between WNV and host defenses both informs basic molecular virology and promotes the development of host- and virus-targeted antiviral strategies.
Keywords: TRIM6; VAMP8; West Nile virus; flavivirus; immunology; type I interferon pathway; ubiquitin.
Copyright © 2020 American Society for Microbiology.
Figures

Similar articles
-
Inhibitor of κB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection.J Biol Chem. 2011 Dec 30;286(52):44412-23. doi: 10.1074/jbc.M111.285205. Epub 2011 Nov 7. J Biol Chem. 2011. PMID: 22065572 Free PMC article.
-
The Matrix Protein of Nipah Virus Targets the E3-Ubiquitin Ligase TRIM6 to Inhibit the IKKε Kinase-Mediated Type-I IFN Antiviral Response.PLoS Pathog. 2016 Sep 13;12(9):e1005880. doi: 10.1371/journal.ppat.1005880. eCollection 2016 Sep. PLoS Pathog. 2016. PMID: 27622505 Free PMC article.
-
The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication.J Virol. 2017 Aug 24;91(18):e00833-17. doi: 10.1128/JVI.00833-17. Print 2017 Sep 15. J Virol. 2017. PMID: 28679761 Free PMC article.
-
The innate immune playbook for restricting West Nile virus infection.Viruses. 2013 Oct 30;5(11):2643-58. doi: 10.3390/v5112643. Viruses. 2013. PMID: 24178712 Free PMC article. Review.
-
Measure and countermeasure: type I IFN (IFN-alpha/beta) antiviral response against West Nile virus.J Innate Immun. 2009;1(5):435-45. doi: 10.1159/000226248. Epub 2009 Jun 24. J Innate Immun. 2009. PMID: 20375601 Free PMC article. Review.
Cited by
-
TRIM Proteins in Host Defense and Viral Pathogenesis.Curr Clin Microbiol Rep. 2020;7(4):101-114. doi: 10.1007/s40588-020-00150-8. Epub 2020 Aug 8. Curr Clin Microbiol Rep. 2020. PMID: 32837832 Free PMC article. Review.
-
Post-fever retinitis - Newer concepts.Indian J Ophthalmol. 2020 Sep;68(9):1775-1786. doi: 10.4103/ijo.IJO_1352_20. Indian J Ophthalmol. 2020. PMID: 32823394 Free PMC article. Review.
-
The RNA helicase DHX16 recognizes specific viral RNA to trigger RIG-I-dependent innate antiviral immunity.Cell Rep. 2022 Mar 8;38(10):110434. doi: 10.1016/j.celrep.2022.110434. Cell Rep. 2022. PMID: 35263596 Free PMC article.
-
Molecular pathogenesis of Japanese encephalitis and possible therapeutic strategies.Arch Virol. 2022 Sep;167(9):1739-1762. doi: 10.1007/s00705-022-05481-z. Epub 2022 Jun 2. Arch Virol. 2022. PMID: 35654913 Free PMC article. Review.
-
Hepatocyte CD36 modulates UBQLN1-mediated proteasomal degradation of autophagic SNARE proteins contributing to septic liver injury.Autophagy. 2023 Sep;19(9):2504-2519. doi: 10.1080/15548627.2023.2196876. Epub 2023 Apr 23. Autophagy. 2023. PMID: 37014234 Free PMC article.
References
-
- Gubler DJ, Kuno G, Markoff L. 2007. Flaviviruses, p 1153–1252. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA.
-
- Lindenbach BD, Thiel HJ, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA.
-
- . 2019. West Nile virus. CDC, Atlanta, GA: https://www.cdc.gov/westnile/index.html.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
