

Long-Chain Acyl-CoA Synthetase 1 Role in Sepsis and Immunity: Perspectives From a Parallel Review of Public Transcriptome Datasets and of the Literature
- PMID: 31681299
- PMCID: PMC6813721
- DOI: 10.3389/fimmu.2019.02410
Long-Chain Acyl-CoA Synthetase 1 Role in Sepsis and Immunity: Perspectives From a Parallel Review of Public Transcriptome Datasets and of the Literature
Abstract
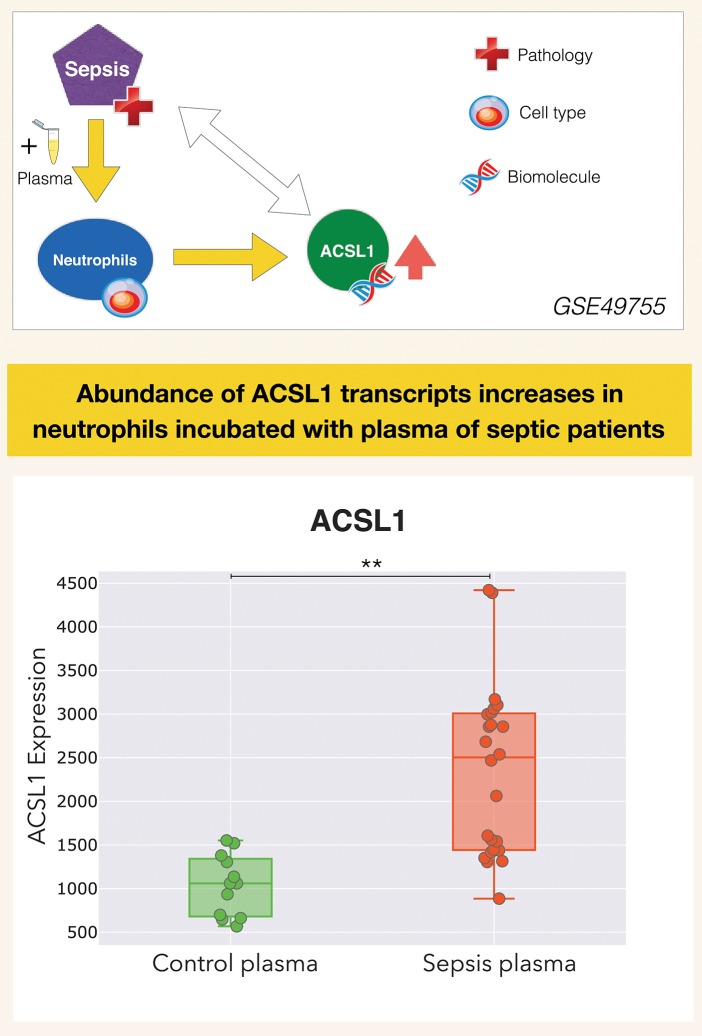
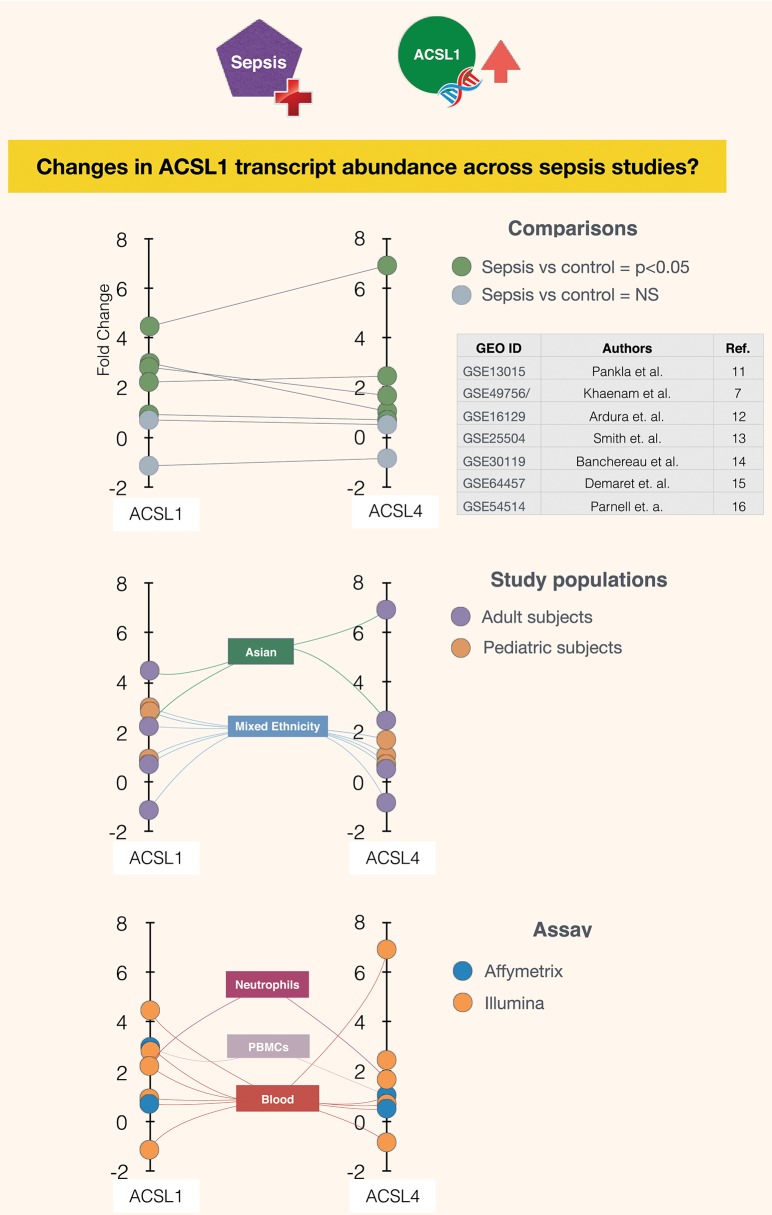
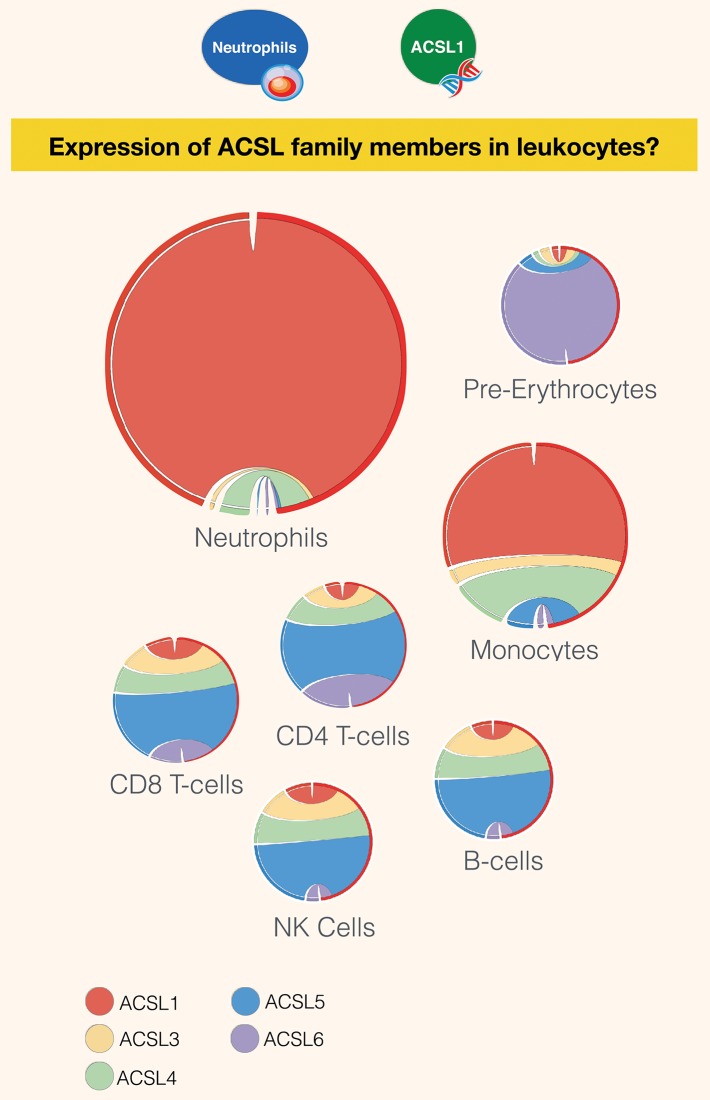
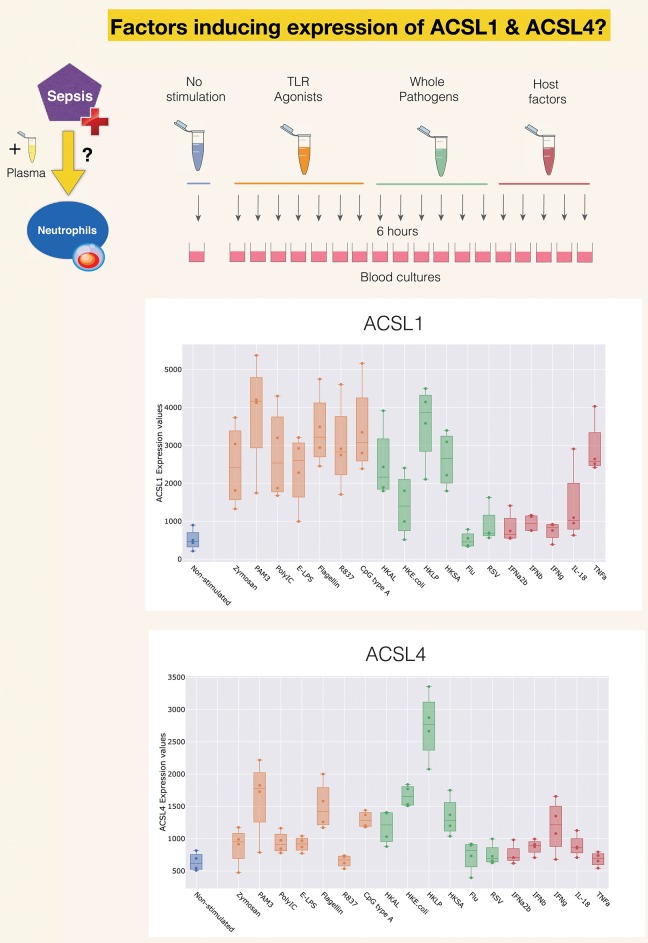
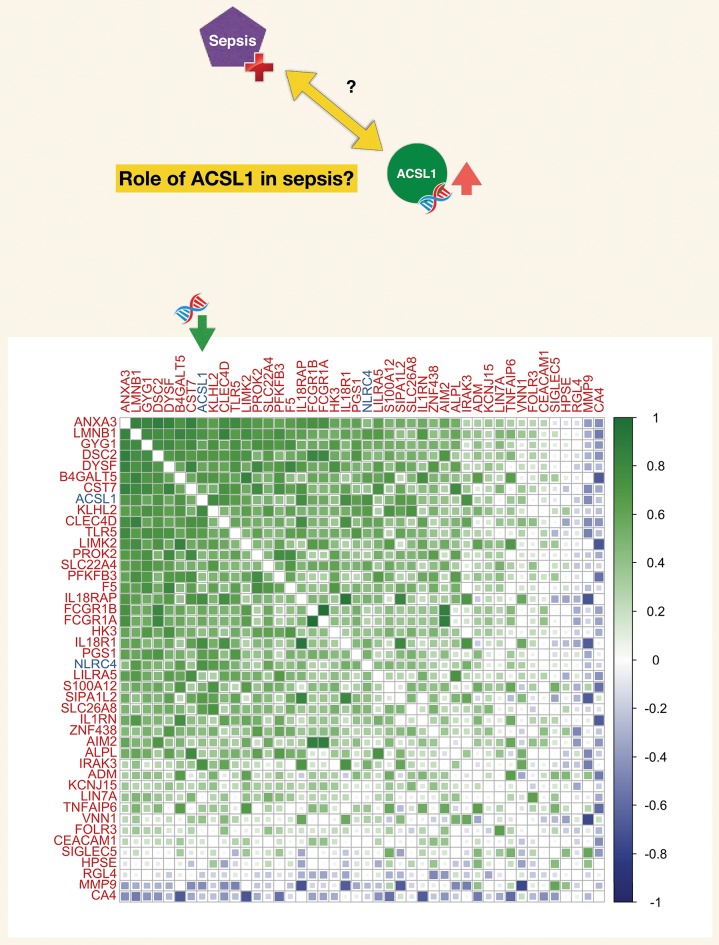
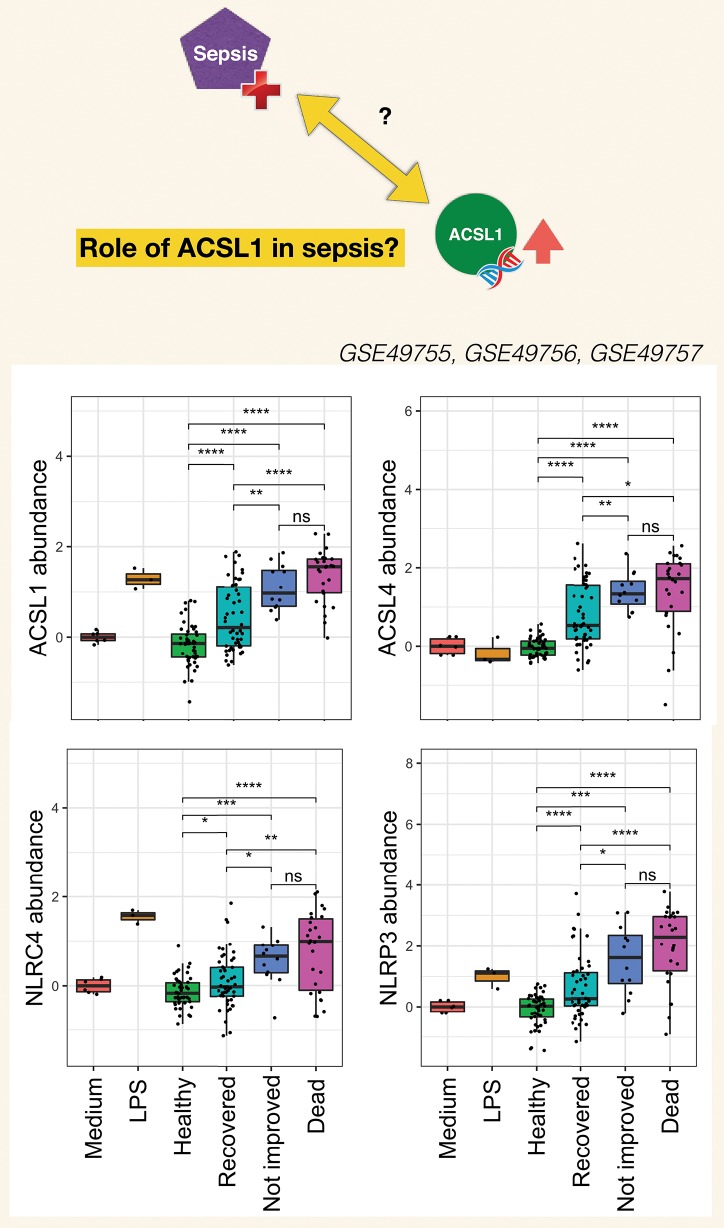
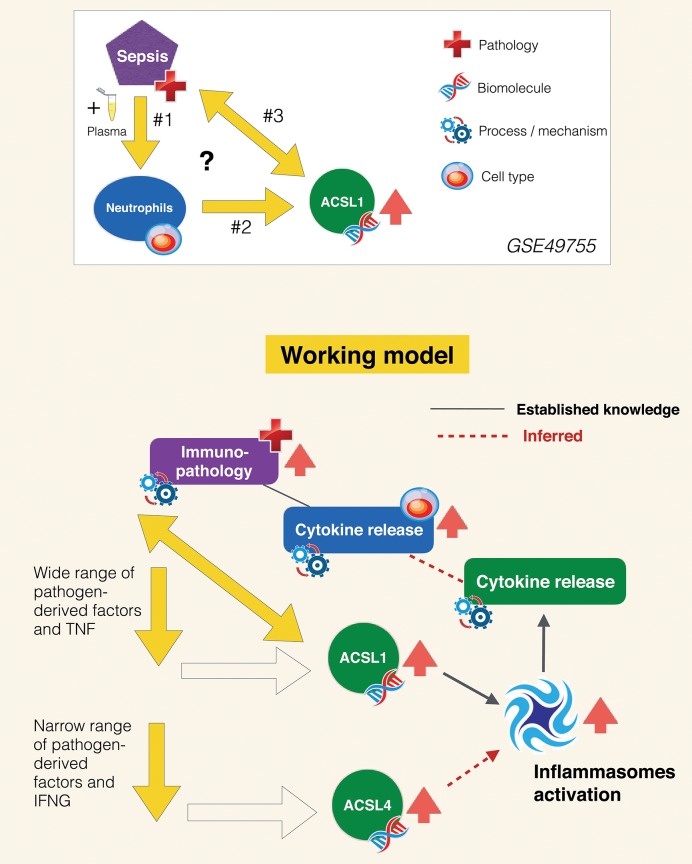
A potential role for the long-chain acyl-CoA synthetase family member 1 (ACSL1) in the immunobiology of sepsis was explored during a hands-on training workshop. Participants first assessed the robustness of the potential gap in biomedical knowledge identified via an initial screen of public transcriptome data and of the literature associated with ACSL1. Increase in ACSL1 transcript abundance during sepsis was confirmed in several independent datasets. Querying the ACSL1 literature also confirmed the absence of reports associating ACSL1 with sepsis. Inferences drawn from both the literature (via indirect associations) and public transcriptome data (via correlation) point to the likely participation of ACSL1 and ACSL4, another family member, in inflammasome activation in neutrophils during sepsis. Furthermore, available clinical data indicate that levels of ACSL1 and ACSL4 induction was significantly higher in fatal cases of sepsis. This denotes potential translational relevance and is consistent with involvement in pathways driving potentially deleterious systemic inflammation. Finally, while ACSL1 expression was induced in blood in vitro by a wide range of pathogen-derived factors as well as TNF, induction of ACSL4 appeared restricted to flagellated bacteria and pathogen-derived TLR5 agonists and IFNG. Taken together, this joint review of public literature and omics data records points to two members of the acyl-CoA synthetase family potentially playing a role in inflammasome activation in neutrophils. Translational relevance of these observations in the context of sepsis and other inflammatory conditions remain to be investigated.
Keywords: OMICS data; lipid metabolism; long-chain acyl-CoA synthetase; neutrophils; sepsis.
Copyright © 2019 Roelands, Garand, Hinchcliff, Ma, Shah, Toufiq, Alfaki, Hendrickx, Boughorbel, Rinchai, Jazaeri, Bedognetti and Chaussabel.
Figures
Similar articles
-
Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking.Circulation. 2019 Jun 11;139(24):2765-2777. doi: 10.1161/CIRCULATIONAHA.119.039610. Epub 2019 Mar 26. Circulation. 2019. PMID: 30909726 Free PMC article.
-
Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy.Mol Cell Biol. 2011 Mar;31(6):1252-62. doi: 10.1128/MCB.01085-10. Epub 2011 Jan 18. Mol Cell Biol. 2011. PMID: 21245374 Free PMC article.
-
Acyl CoA synthetase-1 links facilitated long chain fatty acid uptake to intracellular metabolic trafficking differently in hearts of male versus female mice.J Mol Cell Cardiol. 2016 May;94:1-9. doi: 10.1016/j.yjmcc.2016.03.006. Epub 2016 Mar 16. J Mol Cell Cardiol. 2016. PMID: 26995156 Free PMC article.
-
Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism.Prostaglandins Other Lipid Mediat. 2019 Oct;144:106363. doi: 10.1016/j.prostaglandins.2019.106363. Epub 2019 Jul 12. Prostaglandins Other Lipid Mediat. 2019. PMID: 31306767 Review.
-
Annexin A3 in sepsis: novel perspectives from an exploration of public transcriptome data.Immunology. 2020 Dec;161(4):291-302. doi: 10.1111/imm.13239. Epub 2020 Aug 31. Immunology. 2020. PMID: 32682335 Free PMC article. Review.
Cited by
-
Transcriptional regulation of Acsl1 by CHREBP and NF-kappa B in macrophages during hyperglycemia and inflammation.PLoS One. 2022 Sep 2;17(9):e0272986. doi: 10.1371/journal.pone.0272986. eCollection 2022. PLoS One. 2022. PMID: 36054206 Free PMC article.
-
Transcriptomic profile investigations highlight a putative role for NUDT16 in sepsis.J Cell Mol Med. 2022 Mar;26(5):1714-1721. doi: 10.1111/jcmm.17240. Epub 2022 Feb 17. J Cell Mol Med. 2022. PMID: 35174610 Free PMC article.
-
Identification of toll-like receptor 5 and acyl-CoA synthetase long chain family member 1 as hub genes are correlated with the severe forms of COVID-19 by Weighted gene co-expression network analysis.IET Syst Biol. 2023 Dec;17(6):327-335. doi: 10.1049/syb2.12079. Epub 2023 Oct 12. IET Syst Biol. 2023. PMID: 37823415 Free PMC article.
-
Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection.Viruses. 2021 Nov 28;13(12):2383. doi: 10.3390/v13122383. Viruses. 2021. PMID: 34960652 Free PMC article.
-
The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer.Cancers (Basel). 2024 Mar 16;16(6):1170. doi: 10.3390/cancers16061170. Cancers (Basel). 2024. PMID: 38539505 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous