

Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing
- PMID: 31645721
- PMCID: PMC7253295
- DOI: 10.1038/s41586-019-1658-5
Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing
Abstract
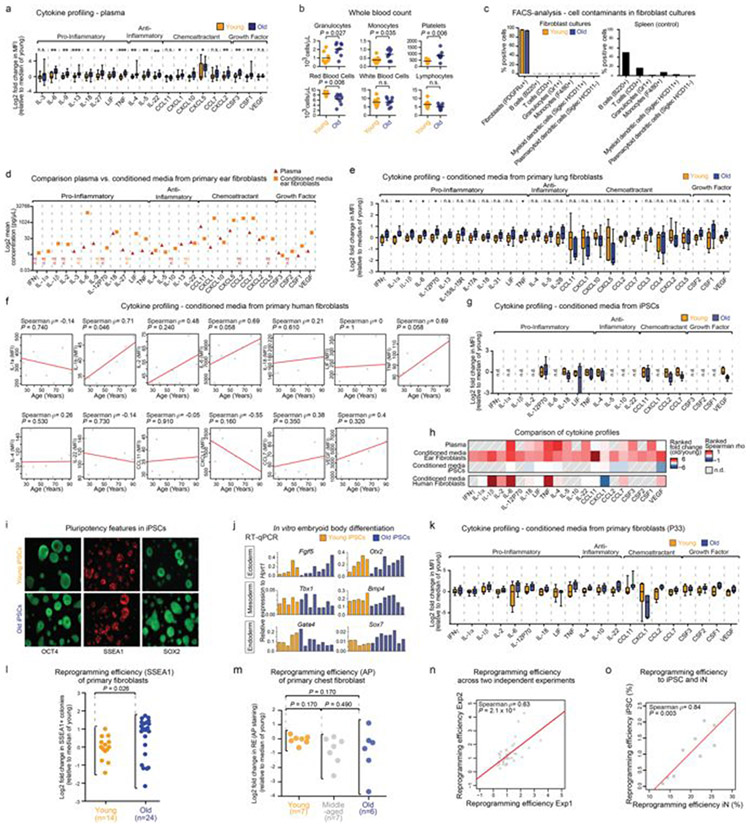
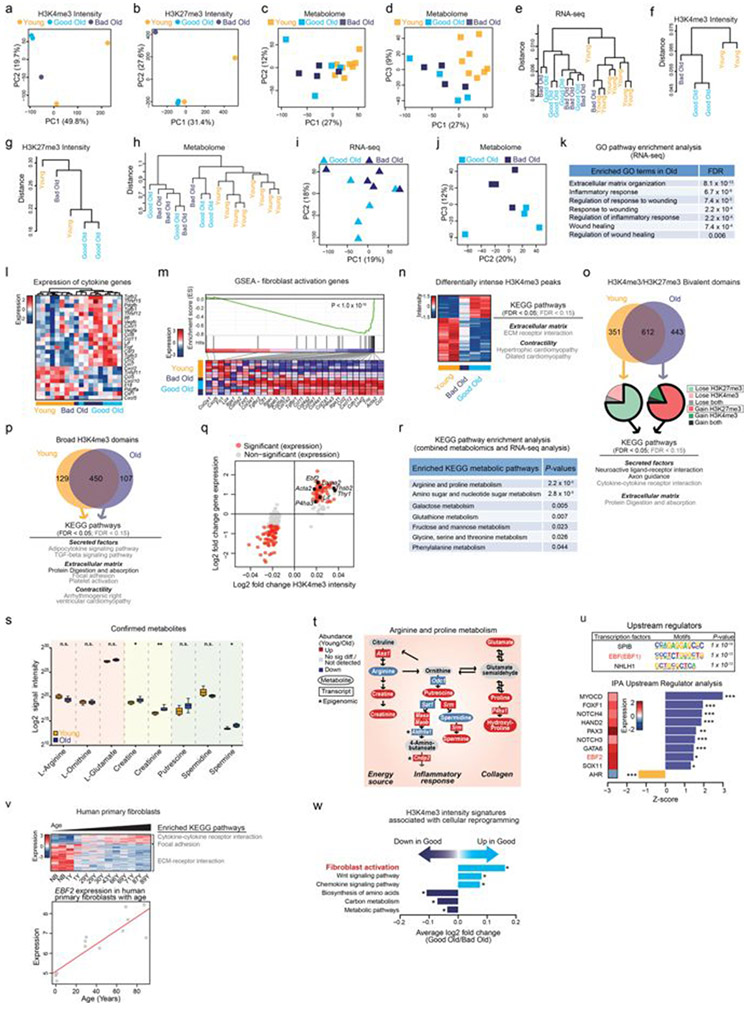
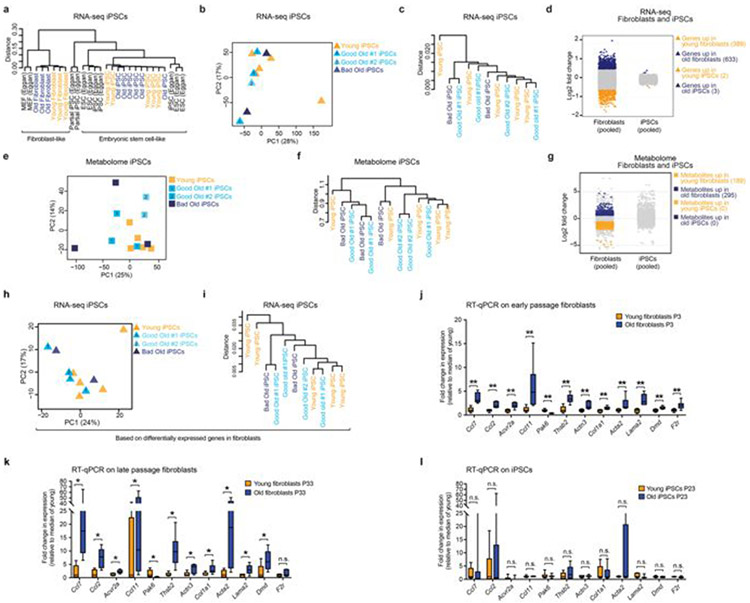
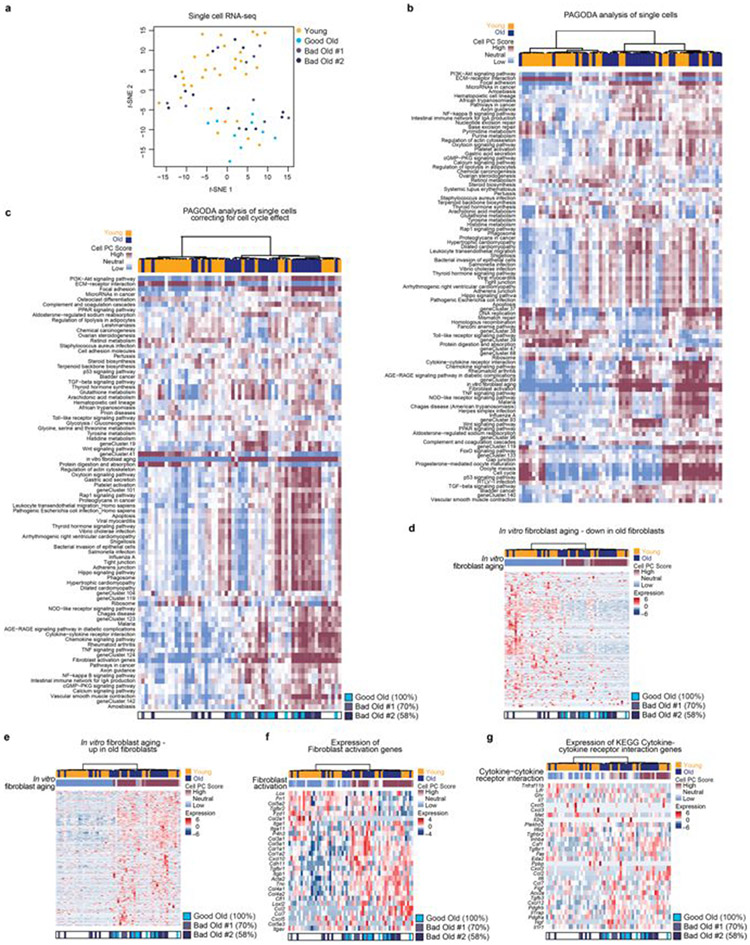
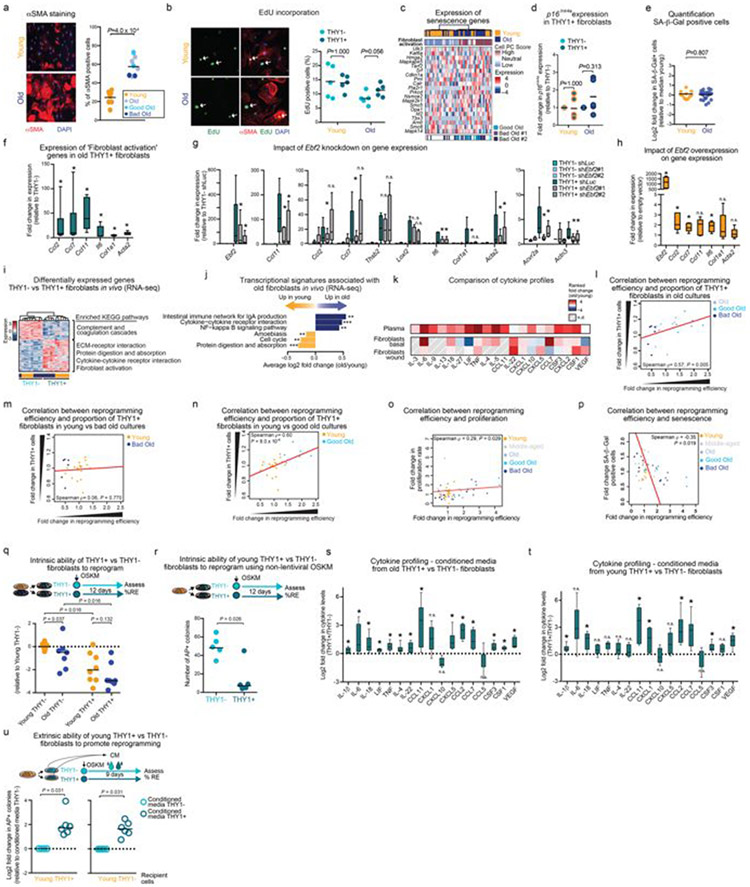
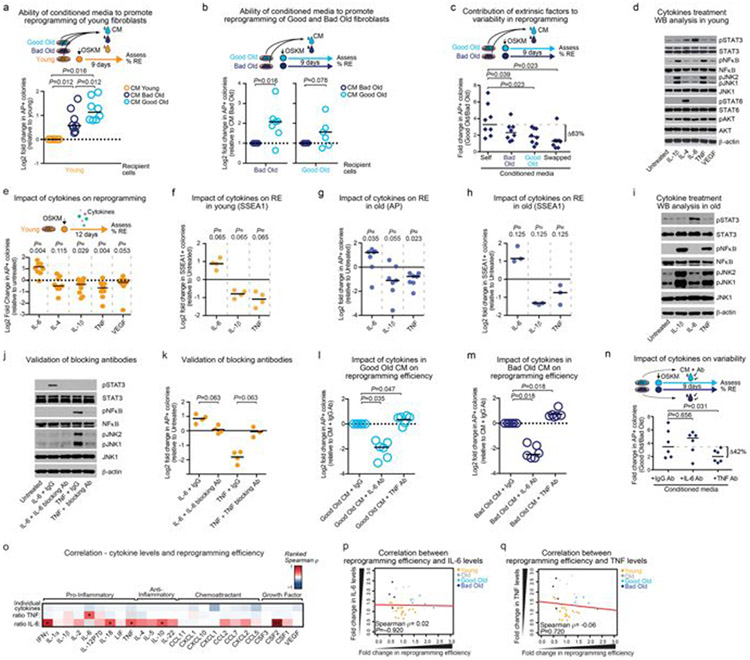
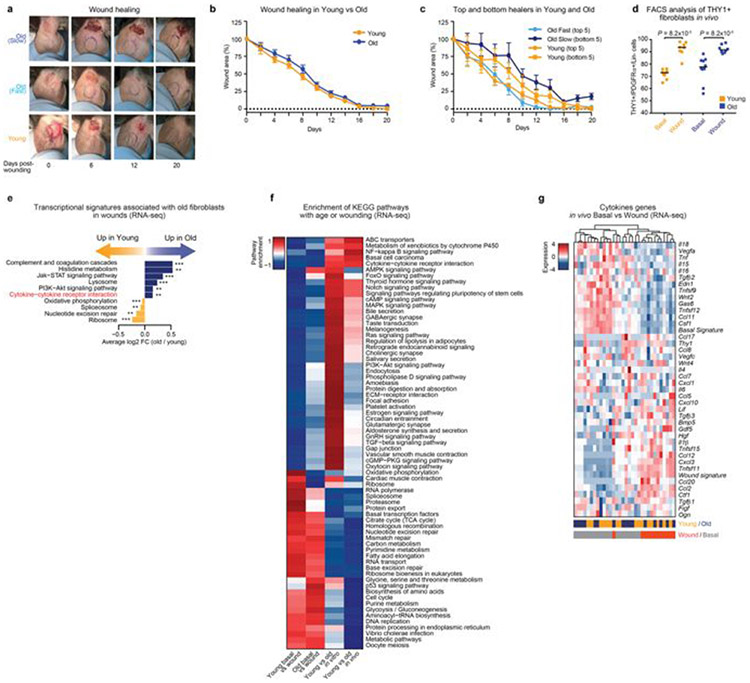
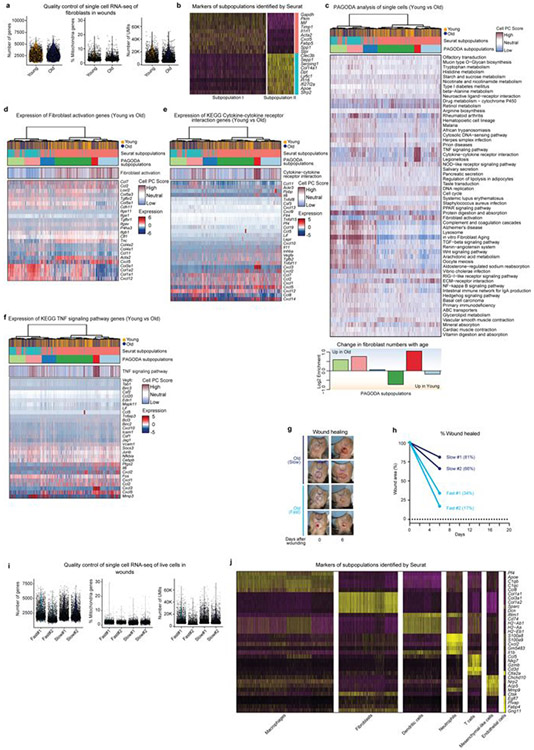
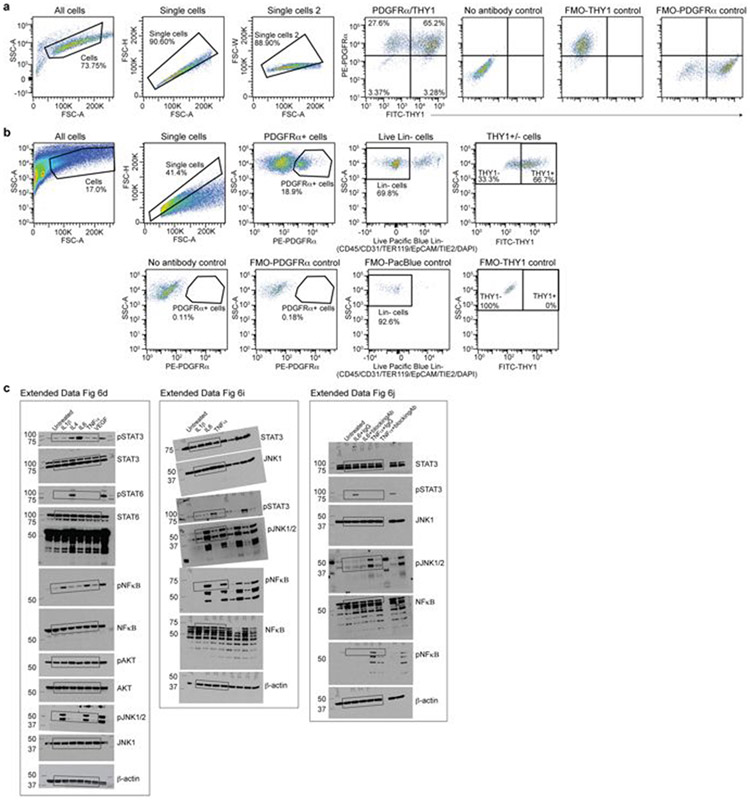
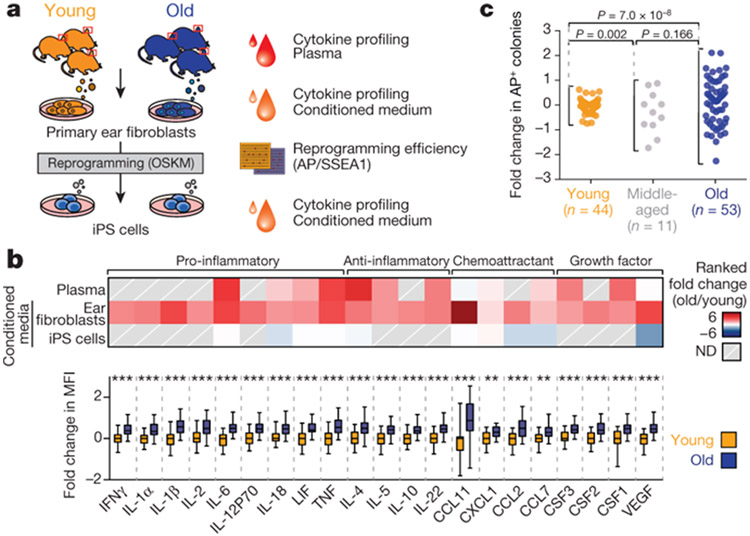
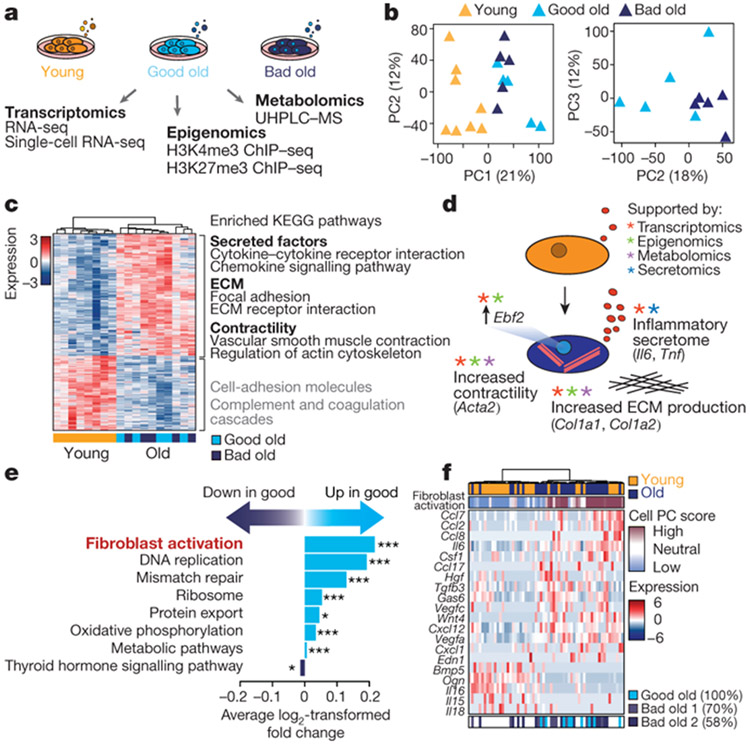
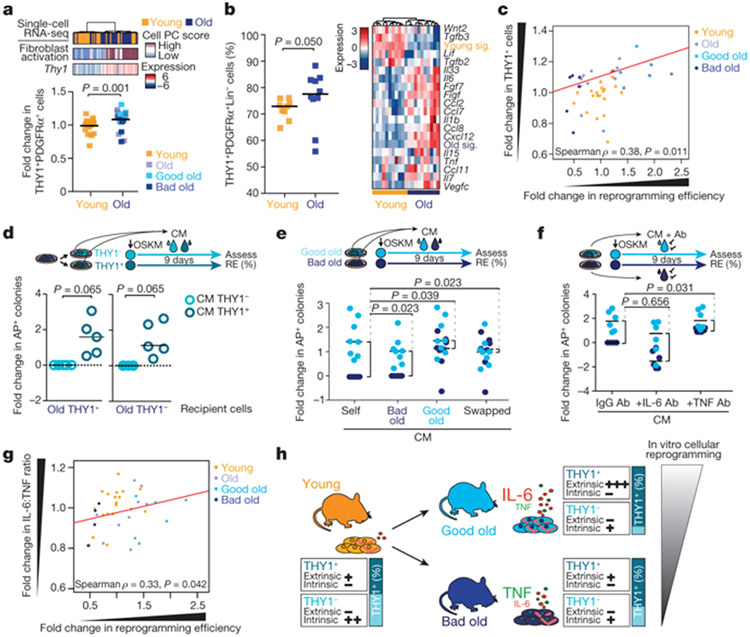
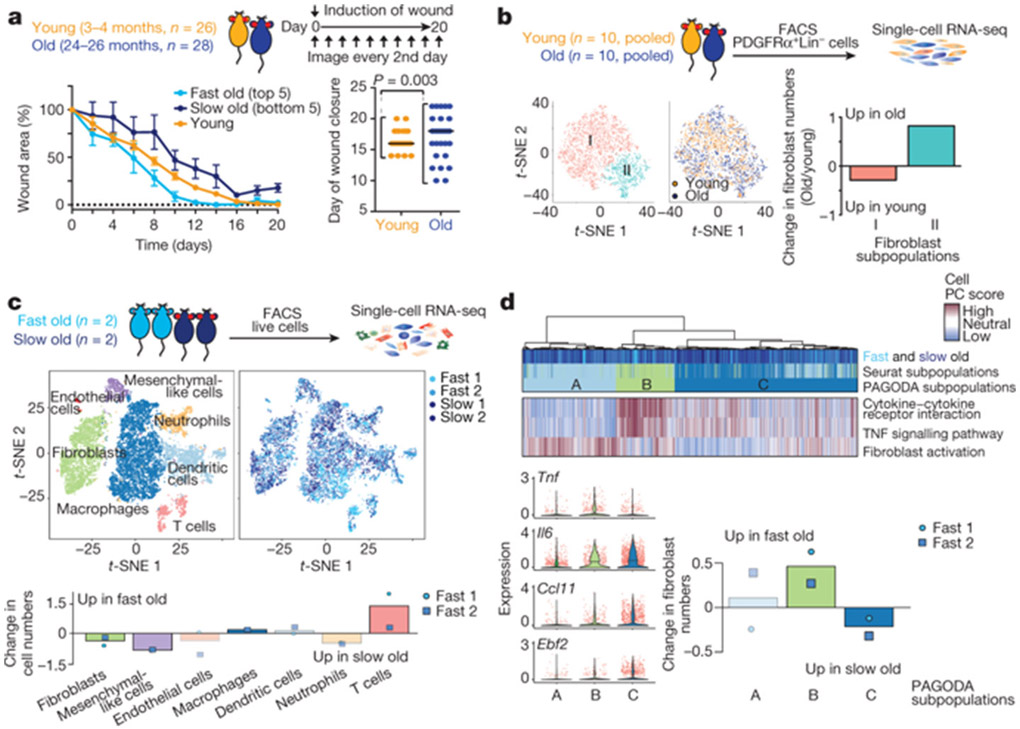
Age-associated chronic inflammation (inflammageing) is a central hallmark of ageing1, but its influence on specific cells remains largely unknown. Fibroblasts are present in most tissues and contribute to wound healing2,3. They are also the most widely used cell type for reprogramming to induced pluripotent stem (iPS) cells, a process that has implications for regenerative medicine and rejuvenation strategies4. Here we show that fibroblast cultures from old mice secrete inflammatory cytokines and exhibit increased variability in the efficiency of iPS cell reprogramming between mice. Variability between individuals is emerging as a feature of old age5-8, but the underlying mechanisms remain unknown. To identify drivers of this variability, we performed multi-omics profiling of fibroblast cultures from young and old mice that have different reprogramming efficiencies. This approach revealed that fibroblast cultures from old mice contain 'activated fibroblasts' that secrete inflammatory cytokines, and that the proportion of activated fibroblasts in a culture correlates with the reprogramming efficiency of that culture. Experiments in which conditioned medium was swapped between cultures showed that extrinsic factors secreted by activated fibroblasts underlie part of the variability between mice in reprogramming efficiency, and we have identified inflammatory cytokines, including TNF, as key contributors. Notably, old mice also exhibited variability in wound healing rate in vivo. Single-cell RNA-sequencing analysis identified distinct subpopulations of fibroblasts with different cytokine expression and signalling in the wounds of old mice with slow versus fast healing rates. Hence, a shift in fibroblast composition, and the ratio of inflammatory cytokines that they secrete, may drive the variability between mice in reprogramming in vitro and influence wound healing rate in vivo. This variability may reflect distinct stochastic ageing trajectories between individuals, and could help in developing personalized strategies to improve iPS cell generation and wound healing in elderly individuals.
Figures

Comment in
-
Ageing is associated with increased variability of cellular reprogramming and wound healing.Cardiovasc Res. 2020 Nov 1;116(13):e171-e174. doi: 10.1093/cvr/cvaa294. Cardiovasc Res. 2020. PMID: 33096562 No abstract available.
Similar articles
-
NF-κB activation impairs somatic cell reprogramming in ageing.Nat Cell Biol. 2015 Aug;17(8):1004-13. doi: 10.1038/ncb3207. Epub 2015 Jul 27. Nat Cell Biol. 2015. Retraction in: Nat Cell Biol. 2019 Mar;21(3):410. doi: 10.1038/s41556-018-0259-0 PMID: 26214134 Retracted.
-
Regeneration of full-thickness skin defects by differentiated adipose-derived stem cells into fibroblast-like cells by fibroblast-conditioned medium.Stem Cell Res Ther. 2017 Apr 20;8(1):92. doi: 10.1186/s13287-017-0520-7. Stem Cell Res Ther. 2017. PMID: 28427476 Free PMC article.
-
Acceleration of somatic cell reprogramming into the induced pluripotent stem cell using a mycosporine-like amino acid, Porphyra 334.Sci Rep. 2020 Feb 28;10(1):3684. doi: 10.1038/s41598-020-60680-5. Sci Rep. 2020. PMID: 32111890 Free PMC article.
-
Turning back time with emerging rejuvenation strategies.Nat Cell Biol. 2019 Jan;21(1):32-43. doi: 10.1038/s41556-018-0206-0. Epub 2019 Jan 2. Nat Cell Biol. 2019. PMID: 30602763 Free PMC article. Review.
-
mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging.Cell Cycle. 2011 Nov 1;10(21):3658-77. doi: 10.4161/cc.10.21.18128. Epub 2011 Nov 1. Cell Cycle. 2011. PMID: 22052357 Review.
Cited by
-
Oral microbial extracellular DNA initiates periodontitis through gingival degradation by fibroblast-derived cathepsin K in mice.Commun Biol. 2022 Sep 14;5(1):962. doi: 10.1038/s42003-022-03896-7. Commun Biol. 2022. PMID: 36104423 Free PMC article.
-
Chinese herb microneedle patch for wound healing.Bioact Mater. 2021 Mar 21;6(10):3507-3514. doi: 10.1016/j.bioactmat.2021.03.023. eCollection 2021 Oct. Bioact Mater. 2021. PMID: 33817424 Free PMC article.
-
A ride through the epigenetic landscape: aging reversal by reprogramming.Geroscience. 2021 Apr;43(2):463-485. doi: 10.1007/s11357-021-00358-6. Epub 2021 Apr 6. Geroscience. 2021. PMID: 33825176 Free PMC article. Review.
-
Wound-Microenvironment Engineering through Advanced-Dressing Bioprinting.Int J Mol Sci. 2022 Mar 4;23(5):2836. doi: 10.3390/ijms23052836. Int J Mol Sci. 2022. PMID: 35269978 Free PMC article.
-
Potential Reasons for Unresponsiveness to Anti-PD1 Immunotherapy in Young Patients with Advanced Melanoma.Life (Basel). 2021 Nov 30;11(12):1318. doi: 10.3390/life11121318. Life (Basel). 2021. PMID: 34947849 Free PMC article. Review.
References
-
- Franceschi C & Campisi J Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci 69, S4–S9 (2014). - PubMed
-
- Ocampo A, Reddy P & Belmonte JCI Anti-aging strategies based on cellular reprogramming. Trends Mol. Med 22, 725–738 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
