

Genome architecture and stability in the Saccharomyces cerevisiae knockout collection
- PMID: 31511699
- PMCID: PMC6774800
- DOI: 10.1038/s41586-019-1549-9
Genome architecture and stability in the Saccharomyces cerevisiae knockout collection
Abstract
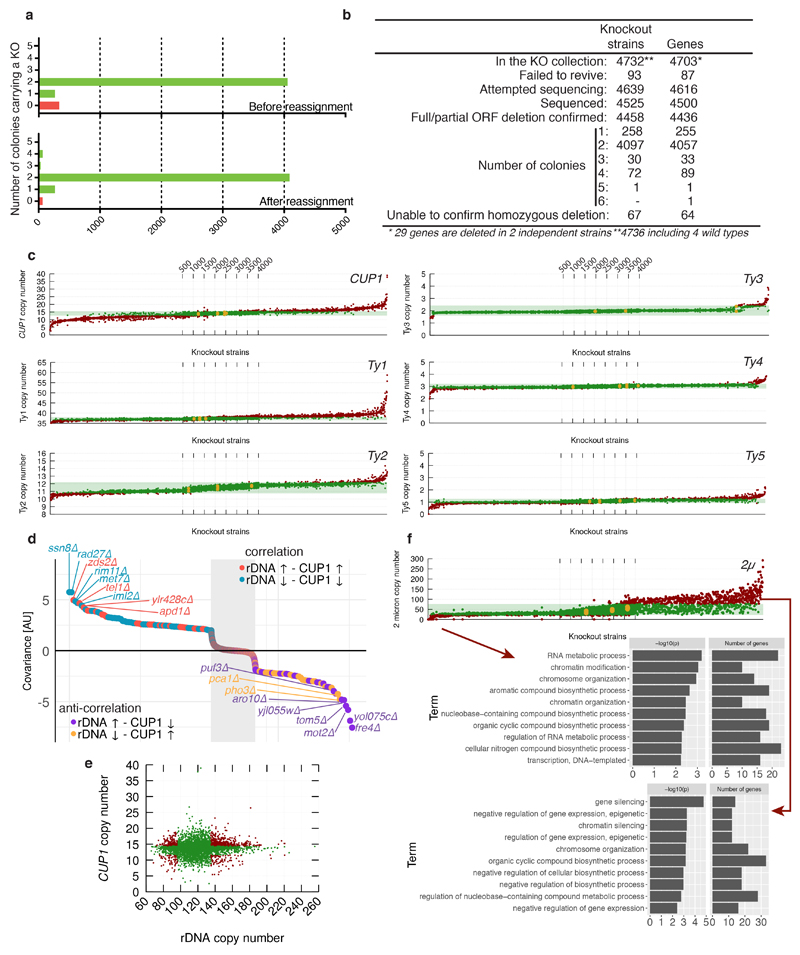
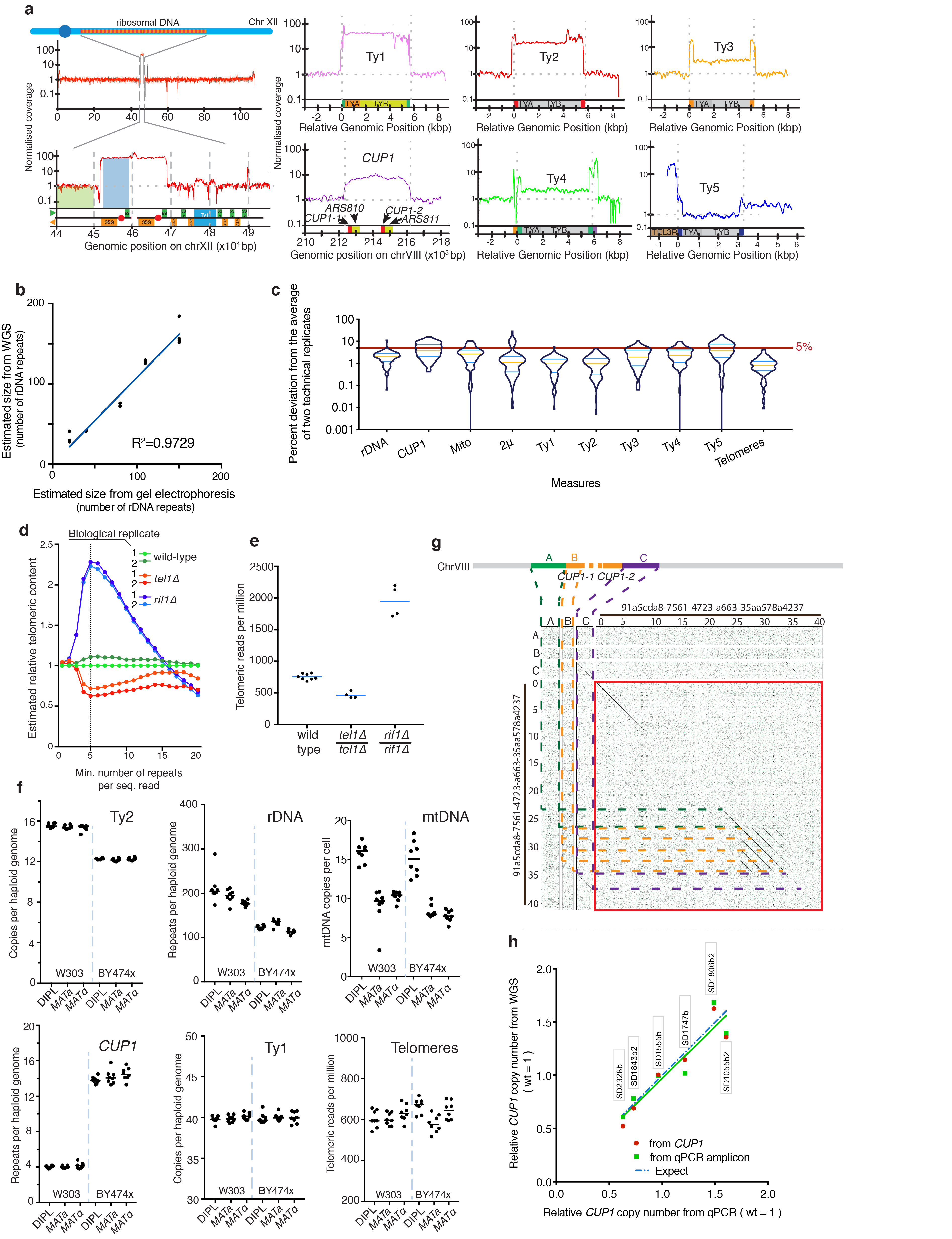
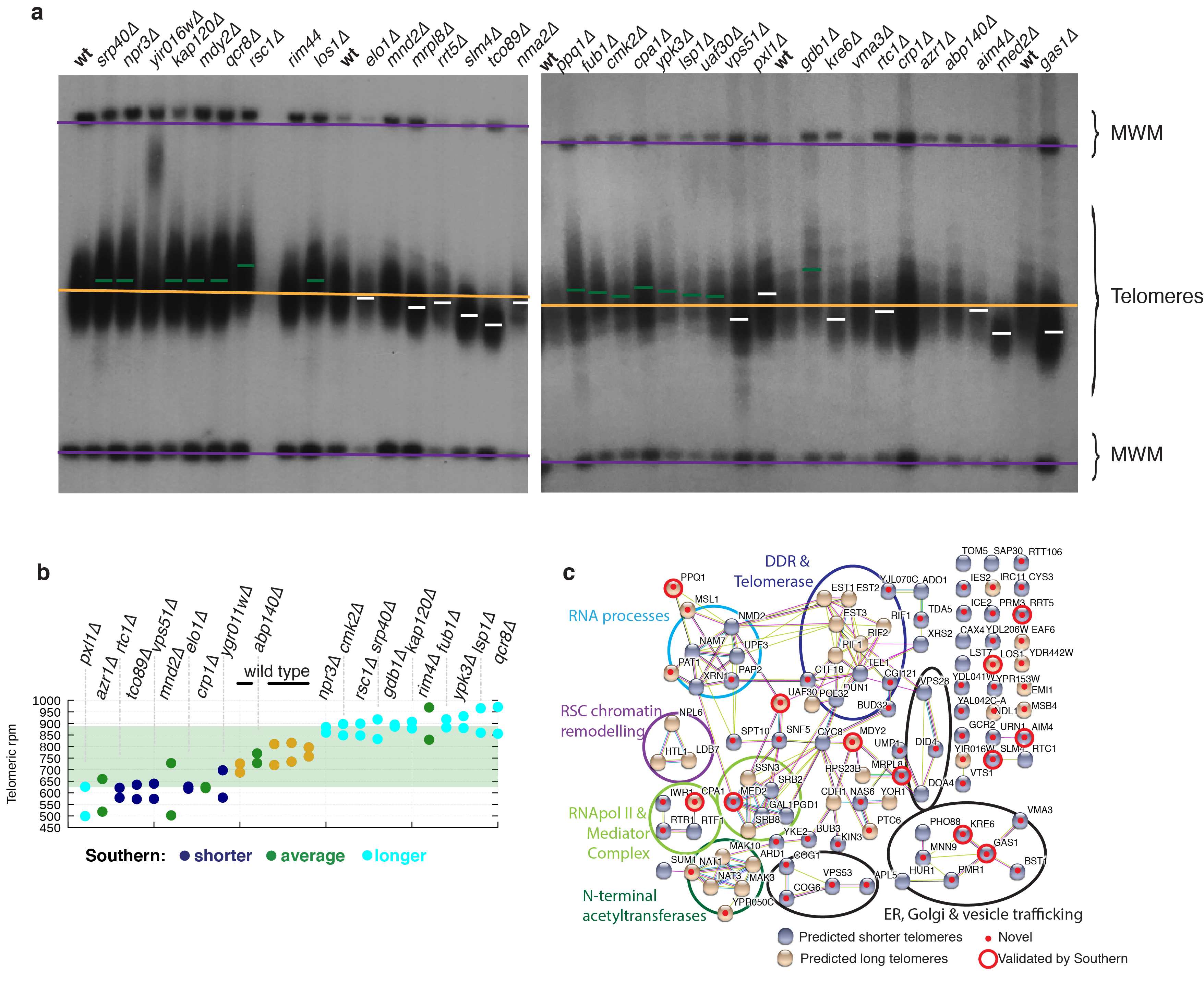
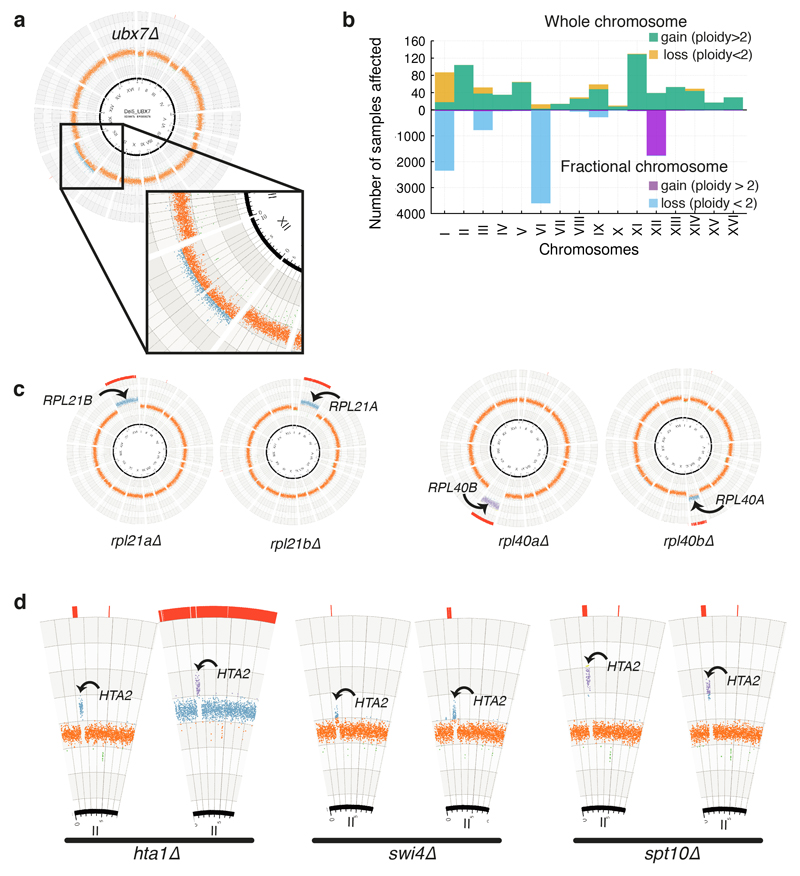
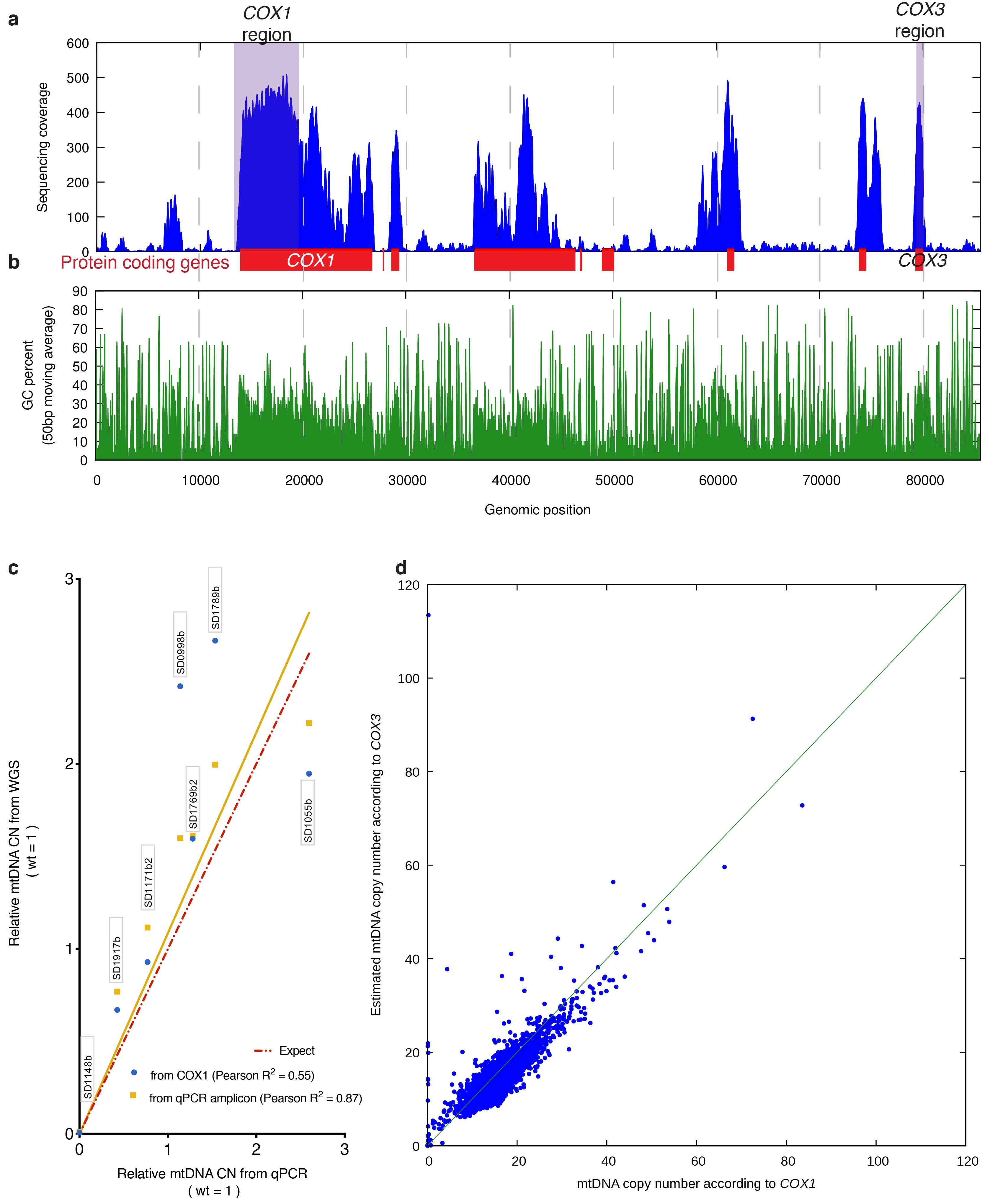
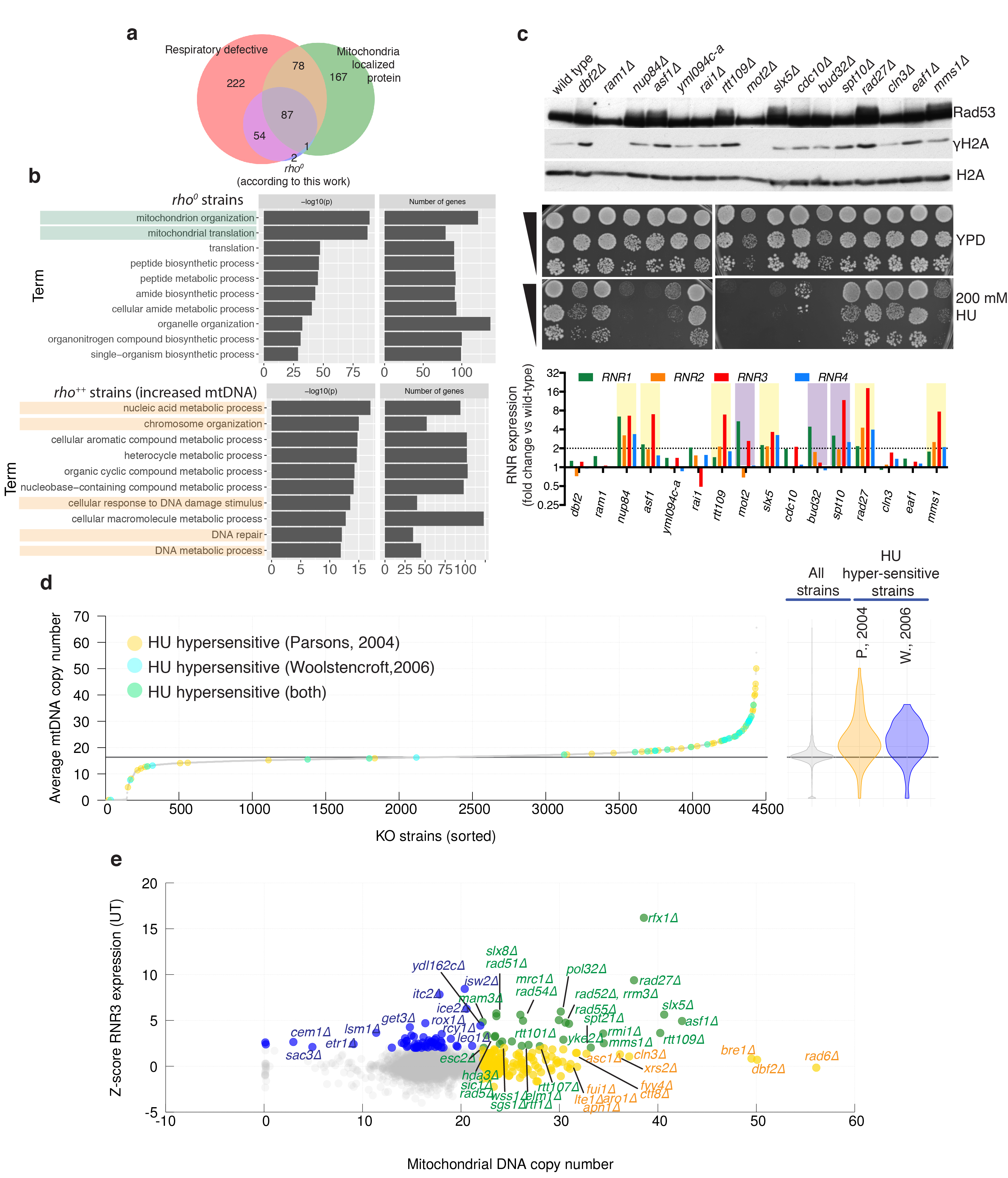
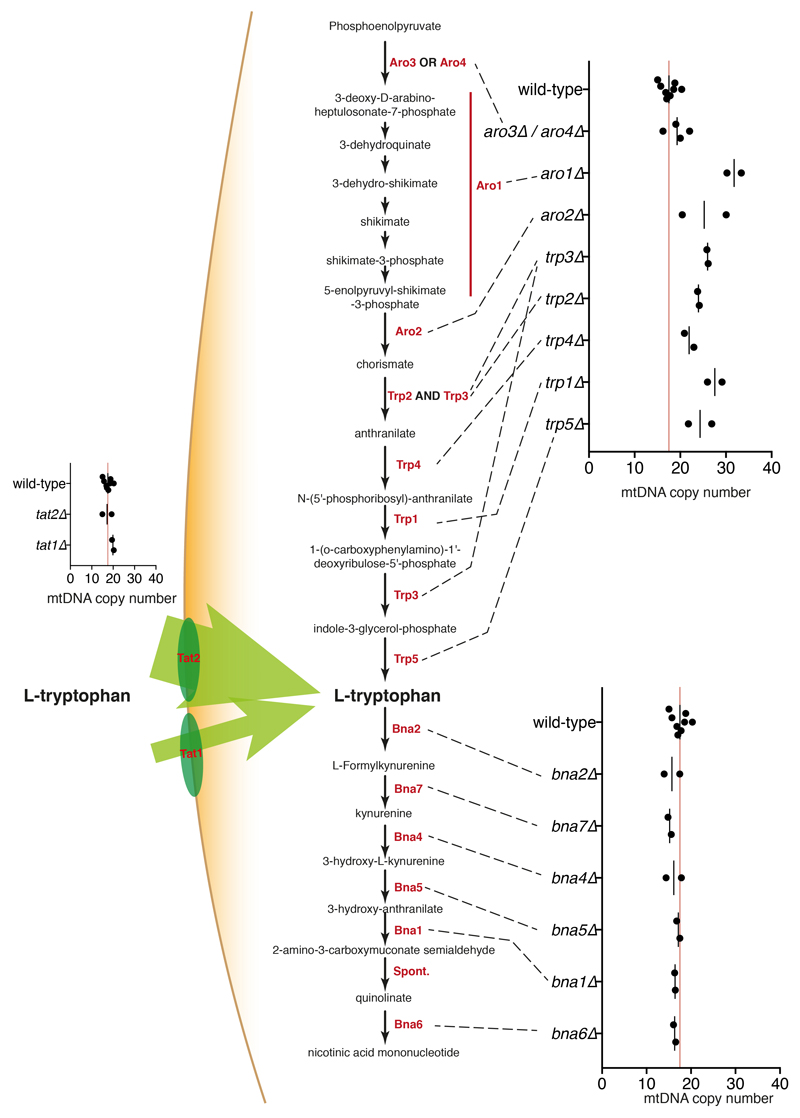
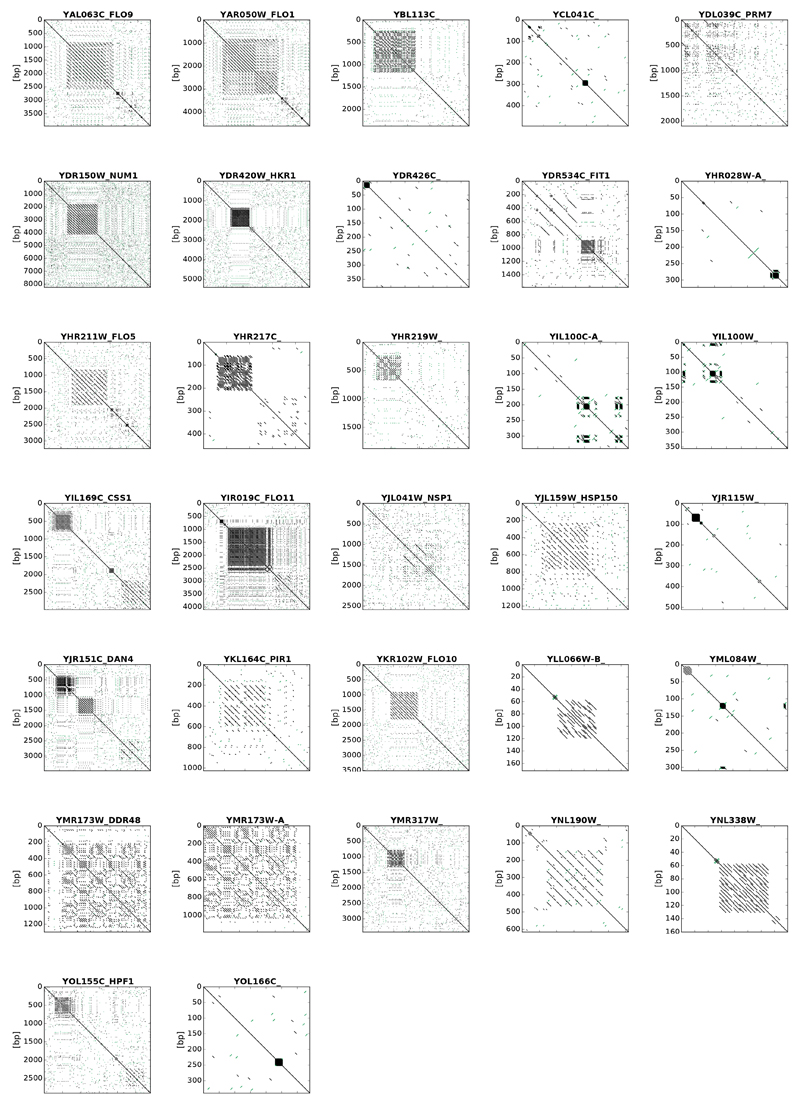
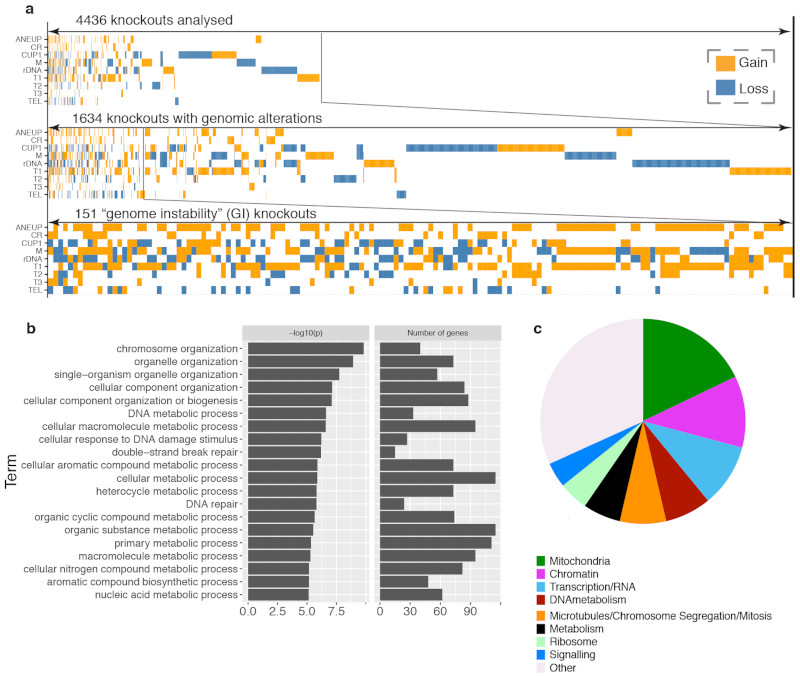
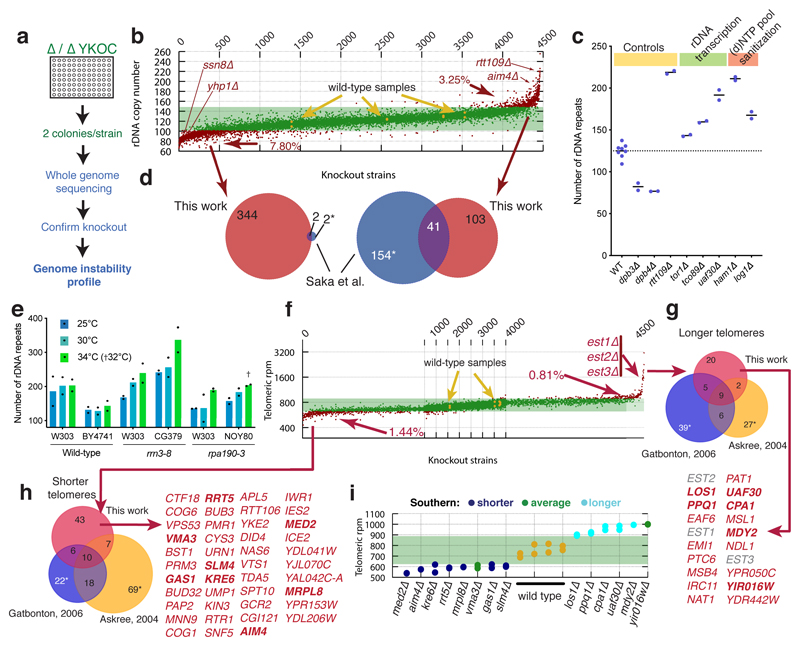
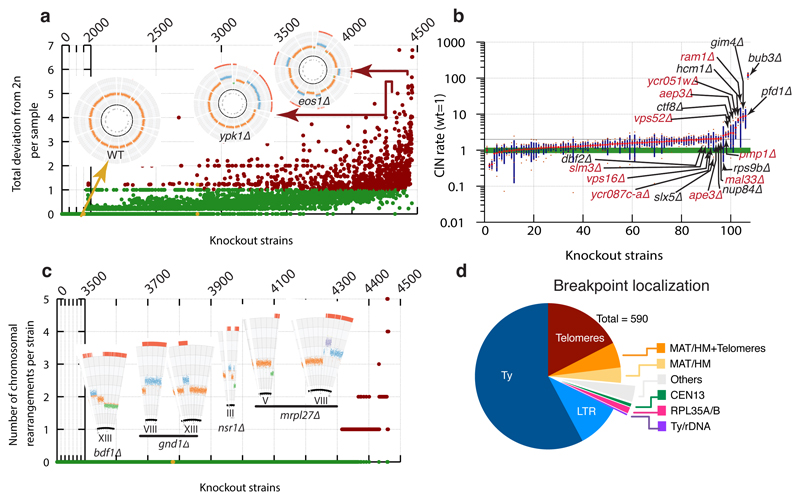
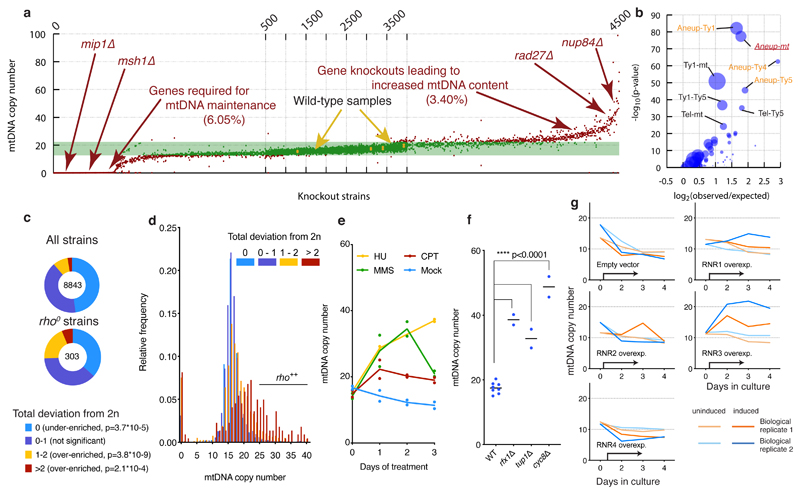
Despite major progress in defining the functional roles of genes, a complete understanding of their influences is far from being realized, even in relatively simple organisms. A major milestone in this direction arose via the completion of the yeast Saccharomyces cerevisiae gene-knockout collection (YKOC), which has enabled high-throughput reverse genetics, phenotypic screenings and analyses of synthetic-genetic interactions1-3. Ensuing experimental work has also highlighted some inconsistencies and mistakes in the YKOC, or genome instability events that rebalance the effects of specific knockouts4-6, but a complete overview of these is lacking. The identification and analysis of genes that are required for maintaining genomic stability have traditionally relied on reporter assays and on the study of deletions of individual genes, but whole-genome-sequencing technologies now enable-in principle-the direct observation of genome instability globally and at scale. To exploit this opportunity, we sequenced the whole genomes of nearly all of the 4,732 strains comprising the homozygous diploid YKOC. Here, by extracting information on copy-number variation of tandem and interspersed repetitive DNA elements, we describe-for almost every single non-essential gene-the genomic alterations that are induced by its loss. Analysis of this dataset reveals genes that affect the maintenance of various genomic elements, highlights cross-talks between nuclear and mitochondrial genome stability, and shows how strains have genetically adapted to life in the absence of individual non-essential genes.
Conflict of interest statement
The authors declare no competing financial or non-financial interests.
Figures

Similar articles
-
Genomic instability in an interspecific hybrid of the genus Saccharomyces: a matter of adaptability.Microb Genom. 2020 Oct;6(10):mgen000448. doi: 10.1099/mgen.0.000448. Microb Genom. 2020. PMID: 33021926 Free PMC article.
-
Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae.Genome Biol. 2009;10(9):R95. doi: 10.1186/gb-2009-10-9-r95. Epub 2009 Sep 14. Genome Biol. 2009. PMID: 19751518 Free PMC article.
-
Diploid-specific [corrected] genome stability genes of S. cerevisiae: genomic screen reveals haploidization as an escape from persisting DNA rearrangement stress.PLoS One. 2011;6(6):e21124. doi: 10.1371/journal.pone.0021124. Epub 2011 Jun 17. PLoS One. 2011. PMID: 21695049 Free PMC article.
-
Navigating yeast genome maintenance with functional genomics.Brief Funct Genomics. 2016 Mar;15(2):119-29. doi: 10.1093/bfgp/elv033. Epub 2015 Aug 31. Brief Funct Genomics. 2016. PMID: 26323482 Review.
-
Ribosomal DNA stability is supported by many 'buffer genes'-introduction to the Yeast rDNA Stability Database.FEMS Yeast Res. 2017 Jan 1;17(1). doi: 10.1093/femsyr/fox001. FEMS Yeast Res. 2017. PMID: 28087673 Review.
Cited by
-
OGG1 inhibition suppresses African swine fever virus replication.Virol Sin. 2023 Feb;38(1):96-107. doi: 10.1016/j.virs.2022.11.006. Epub 2022 Nov 23. Virol Sin. 2023. PMID: 36435451 Free PMC article.
-
Biocontainment strategies for in vivo applications of Saccharomyces boulardii.Front Bioeng Biotechnol. 2023 Feb 20;11:1136095. doi: 10.3389/fbioe.2023.1136095. eCollection 2023. Front Bioeng Biotechnol. 2023. PMID: 36890914 Free PMC article.
-
The proteomic landscape of genome-wide genetic perturbations.Cell. 2023 Apr 27;186(9):2018-2034.e21. doi: 10.1016/j.cell.2023.03.026. Epub 2023 Apr 19. Cell. 2023. PMID: 37080200 Free PMC article.
-
Indole-3-acetic acid is a physiological inhibitor of TORC1 in yeast.PLoS Genet. 2021 Mar 9;17(3):e1009414. doi: 10.1371/journal.pgen.1009414. eCollection 2021 Mar. PLoS Genet. 2021. PMID: 33690632 Free PMC article.
-
Polymorphism of Saccharomyces cerevisiae Strains in DNA Metabolism Genes.Int J Mol Sci. 2023 Apr 25;24(9):7795. doi: 10.3390/ijms24097795. Int J Mol Sci. 2023. PMID: 37175502 Free PMC article.
References
-
- Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science (New York, N.Y.) 2001;294:2364–2368. - PubMed
-
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. - PubMed
-
- Hughes TR, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nature genetics. 2000;25:333–337. - PubMed
-
- Lehner KR, Stone MM, Farber RA, Petes TD. Ninety-six haploid yeast strains with individual disruptions of open reading frames between YOR097C and YOR192C, constructed for the Saccharomyces genome deletion project, have an additional mutation in the mismatch repair gene MSH3. Genetics. 2007;177:1951–1953. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
