

An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity
- PMID: 31391303
- PMCID: PMC6717285
- DOI: 10.1073/pnas.1907031116
An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity
Abstract
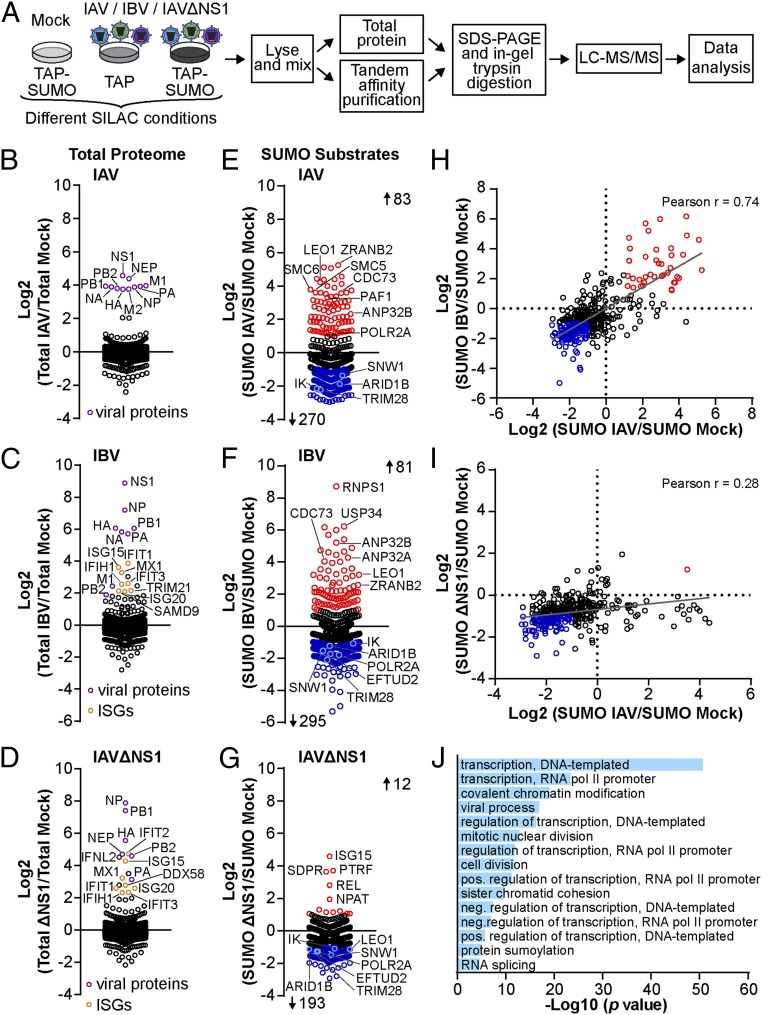
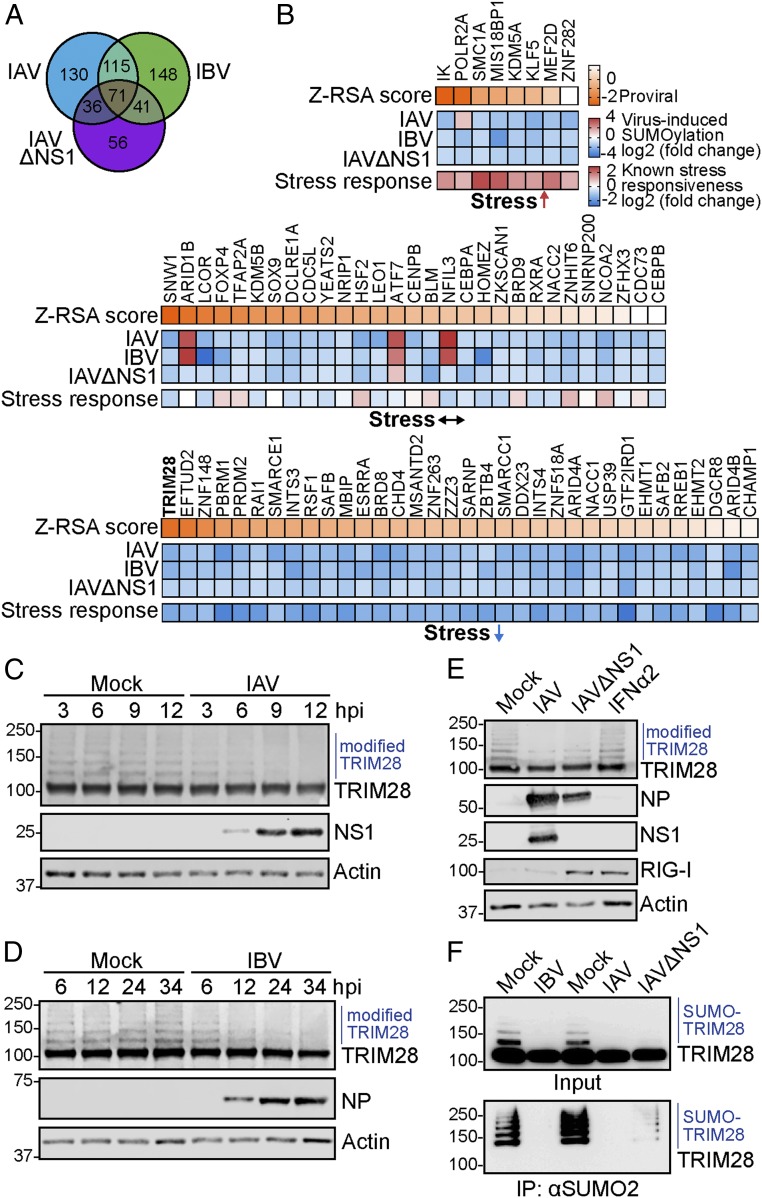
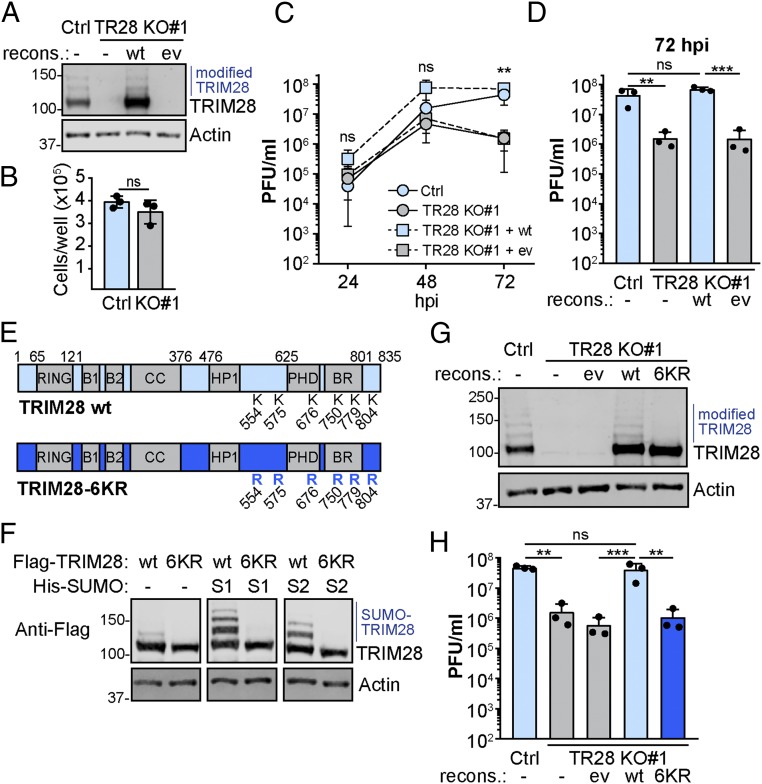
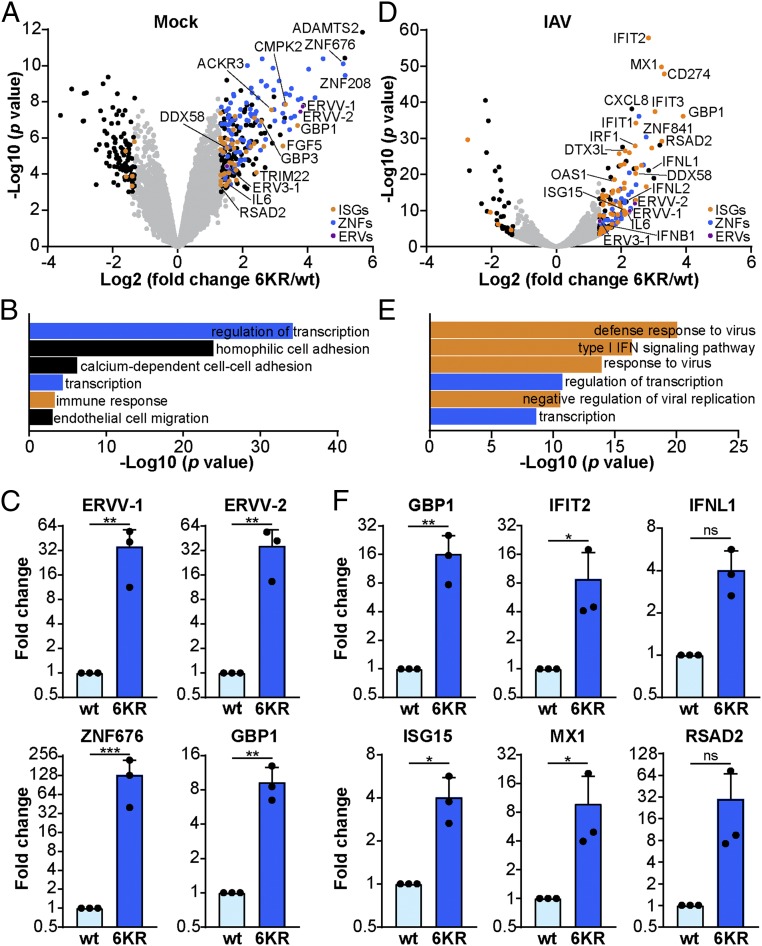
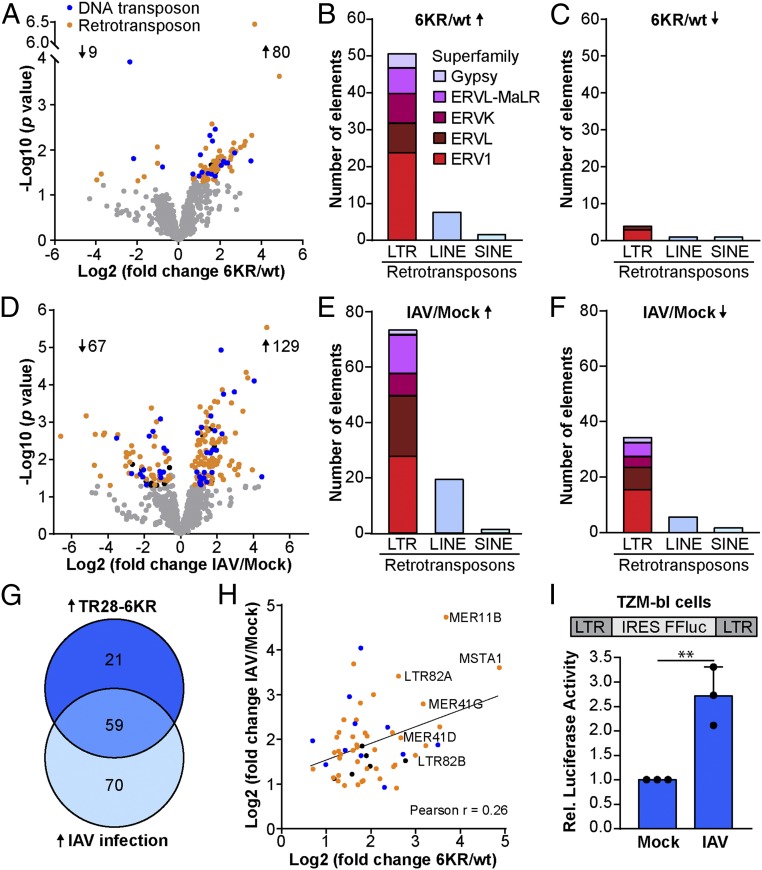
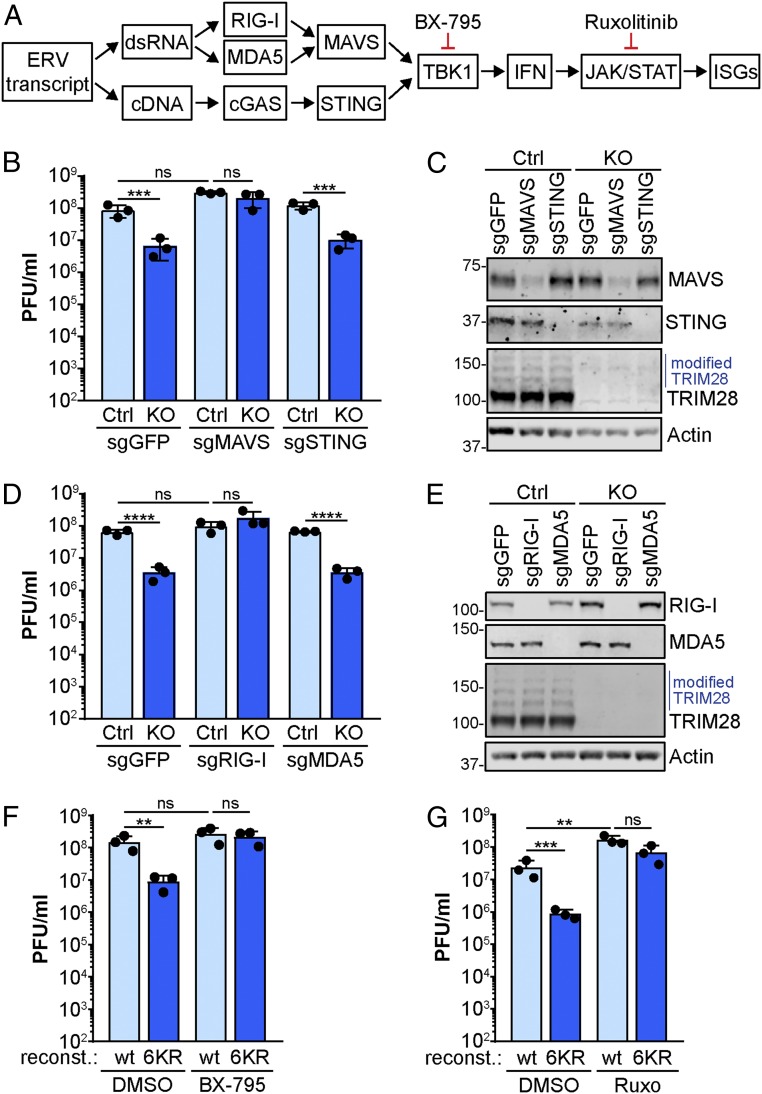
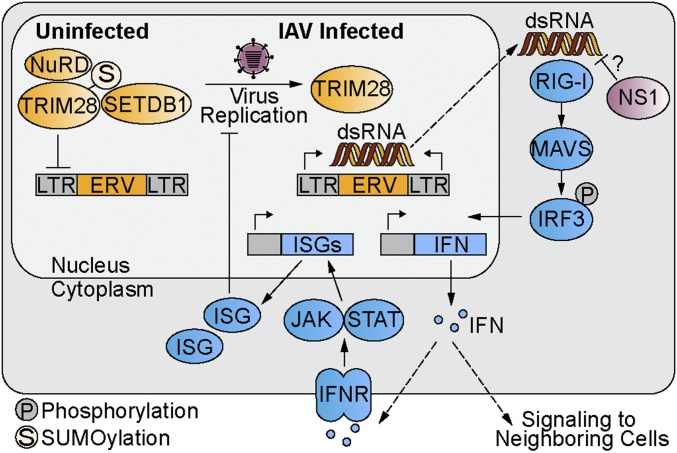
Dynamic small ubiquitin-like modifier (SUMO) linkages to diverse cellular protein groups are critical to orchestrate resolution of stresses such as genome damage, hypoxia, or proteotoxicity. Defense against pathogen insult (often reliant upon host recognition of "non-self" nucleic acids) is also modulated by SUMO, but the underlying mechanisms are incompletely understood. Here, we used quantitative SILAC-based proteomics to survey pan-viral host SUMOylation responses, creating a resource of almost 600 common and unique SUMO remodeling events that are mounted during influenza A and B virus infections, as well as during viral innate immune stimulation. Subsequent mechanistic profiling focused on a common infection-induced loss of the SUMO-modified form of TRIM28/KAP1, a host transcriptional repressor. By integrating knockout and reconstitution models with system-wide transcriptomics, we provide evidence that influenza virus-triggered loss of SUMO-modified TRIM28 leads to derepression of endogenous retroviral (ERV) elements, unmasking this cellular source of "self" double-stranded (ds)RNA. Consequently, loss of SUMO-modified TRIM28 potentiates canonical cytosolic dsRNA-activated IFN-mediated defenses that rely on RIG-I, MAVS, TBK1, and JAK1. Intriguingly, although wild-type influenza A virus robustly triggers this SUMO switch in TRIM28, the induction of IFN-stimulated genes is limited unless expression of the viral dsRNA-binding protein NS1 is abrogated. This may imply a viral strategy to antagonize such a host response by sequestration of induced immunostimulatory ERV dsRNAs. Overall, our data reveal that a key nuclear mechanism that normally prevents aberrant expression of ERV elements (ERVs) has been functionally co-opted via a stress-induced SUMO switch to augment antiviral immunity.
Keywords: SUMO; dsRNA; endogenous retroviruses; influenza; interferon.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Proteomic Approaches to Dissect Host SUMOylation during Innate Antiviral Immune Responses.Viruses. 2021 Mar 23;13(3):528. doi: 10.3390/v13030528. Viruses. 2021. PMID: 33806893 Free PMC article. Review.
-
Novel Role for Protein Inhibitor of Activated STAT 4 (PIAS4) in the Restriction of Herpes Simplex Virus 1 by the Cellular Intrinsic Antiviral Immune Response.J Virol. 2016 Apr 14;90(9):4807-4826. doi: 10.1128/JVI.03055-15. Print 2016 May. J Virol. 2016. PMID: 26937035 Free PMC article.
-
Phosphorylation of TRIM28 Enhances the Expression of IFN-β and Proinflammatory Cytokines During HPAIV Infection of Human Lung Epithelial Cells.Front Immunol. 2018 Sep 28;9:2229. doi: 10.3389/fimmu.2018.02229. eCollection 2018. Front Immunol. 2018. PMID: 30323812 Free PMC article.
-
SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1.J Virol. 2016 Jun 10;90(13):5939-5952. doi: 10.1128/JVI.00426-16. Print 2016 Jul 1. J Virol. 2016. PMID: 27099310 Free PMC article.
-
Viruses, SUMO, and immunity: the interplay between viruses and the host SUMOylation system.J Neurovirol. 2021 Aug;27(4):531-541. doi: 10.1007/s13365-021-00995-9. Epub 2021 Aug 3. J Neurovirol. 2021. PMID: 34342851 Free PMC article. Review.
Cited by
-
Current and prospective control strategies of influenza A virus in swine.Porcine Health Manag. 2021 Feb 28;7(1):23. doi: 10.1186/s40813-021-00196-0. Porcine Health Manag. 2021. PMID: 33648602 Free PMC article. Review.
-
Influenza A Virus Infection Reactivates Human Endogenous Retroviruses Associated with Modulation of Antiviral Immunity.Viruses. 2022 Jul 21;14(7):1591. doi: 10.3390/v14071591. Viruses. 2022. PMID: 35891571 Free PMC article.
-
TRIM28 facilitates type I interferon activation by targeting TBK1.Front Immunol. 2024 Mar 1;15:1279920. doi: 10.3389/fimmu.2024.1279920. eCollection 2024. Front Immunol. 2024. PMID: 38495890 Free PMC article.
-
Activation of cytosolic RNA sensors by endogenous ligands: roles in disease pathogenesis.Front Immunol. 2023 May 24;14:1092790. doi: 10.3389/fimmu.2023.1092790. eCollection 2023. Front Immunol. 2023. PMID: 37292201 Free PMC article. Review.
-
Human Endogenous Retroviruses in Diseases.Subcell Biochem. 2023;106:403-439. doi: 10.1007/978-3-031-40086-5_15. Subcell Biochem. 2023. PMID: 38159236
References
-
- Hu M. M., Shu H. B., Cytoplasmic mechanisms of recognition and defense of microbial nucleic acids. Annu. Rev. Cell Dev. Biol. 34, 357–379 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
