

doi: 10.1038/s41598-019-45839-z.
The ENCODE Blacklist: Identification of Problematic Regions of the Genome
Affiliations
- PMID: 31249361
- PMCID: PMC6597582
- DOI: 10.1038/s41598-019-45839-z
Item in Clipboard
The ENCODE Blacklist: Identification of Problematic Regions of the Genome
Sci Rep.
.
Abstract
Functional genomics assays based on high-throughput sequencing greatly expand our ability to understand the genome. Here, we define the ENCODE blacklist- a comprehensive set of regions in the human, mouse, worm, and fly genomes that have anomalous, unstructured, or high signal in next-generation sequencing experiments independent of cell line or experiment. The removal of the ENCODE blacklist is an essential quality measure when analyzing functional genomics data.
Conflict of interest statement
The authors declare no competing interests.
Figures
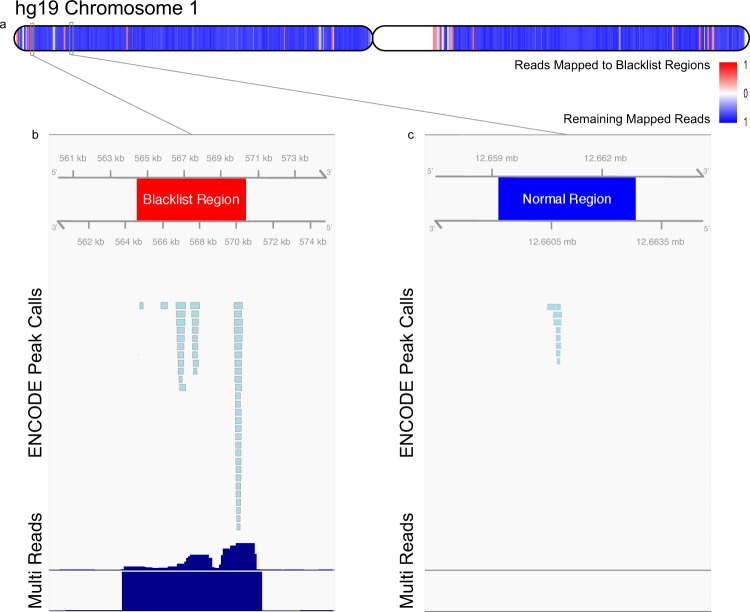
Blacklist regions are tightly distributed across the chromosome and sequester high read mapping signals. (a) Distribution of mapped reads along human chromosome 1 in hg19. (b) An example blacklisted region on chromosome 1. Displayed are pre-filtered ENCODE ChIP-seq peak calls, quantile normalized median read signal (Reads), and quantile normalized median multimapped read signal (Multi). Axes are scaled for illustrative purposes and signal values are truncated at approximately 10-fold enrichment. Signal in these regions are up to 6400× background levels. (c) An example “normal” ENCODE ChIP-seq peak region on chromosome 1 selected as a region containing ChIP-seq peaks.
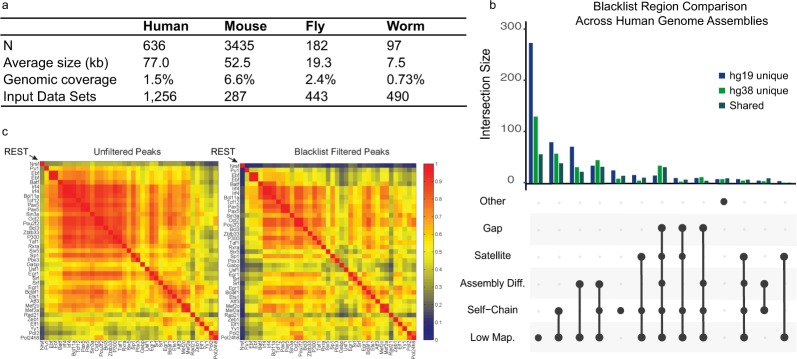
Blacklist regions account for a significant portion of ChIP-seq reads, are driven by artifacts in genome assemblies, and removal of these regions is essential to removing noise in genomics assays. (a) The number of blacklisted regions across species with their average size, genomic coverage, and input datasets excluding assembly gaps used for hg38, mm10, dm6, and ce11 respectively. (b) An UpSet plot displaying the breakdown of uniquely annotated regions in hg19 and hg38, and the shared regions between them. Low-mappability (Low-Map.) regions account for the majority of unique regions in both hg19 and hg38. (c) Applying the blacklist to ChIP-seq peaks results in an overall reduced correlation and, in the highlighted example, results in a more biologically meaningful interpretation of the data.
Similar articles
-
TAG Sequence Identification of Genomic Regions Using TAGdb.Methods Mol Biol. 2016;1374:233-40. doi: 10.1007/978-1-4939-3167-5_12. Methods Mol Biol. 2016. PMID: 26519409
-
DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly.Genome Res. 2014 Dec;24(12):2022-32. doi: 10.1101/gr.175141.114. Epub 2014 Sep 18. Genome Res. 2014. PMID: 25236618 Free PMC article.
-
Pan-Genome Storage and Analysis Techniques.Methods Mol Biol. 2018;1704:29-53. doi: 10.1007/978-1-4939-7463-4_2. Methods Mol Biol. 2018. PMID: 29277862 Review.
-
Genome assembly reborn: recent computational challenges.Brief Bioinform. 2009 Jul;10(4):354-66. doi: 10.1093/bib/bbp026. Epub 2009 May 29. Brief Bioinform. 2009. PMID: 19482960 Free PMC article.
-
An introduction to the informatics of "next-generation" sequencing.Curr Protoc Bioinformatics. 2011 Dec;Chapter 11:11.1.1-11.1.9. doi: 10.1002/0471250953.bi1101s36. Curr Protoc Bioinformatics. 2011. PMID: 22161566 Review.
Cited by
-
Transcriptional programming mediated by the histone demethylase KDM5C regulates dendritic cell population heterogeneity and function.Cell Rep. 2024 Aug 27;43(8):114506. doi: 10.1016/j.celrep.2024.114506. Epub 2024 Jul 24. Cell Rep. 2024. PMID: 39052479 Free PMC article.
-
Functional heterogeneity of alveolar macrophage population based on expression of CXCL2.Sci Immunol. 2020 Aug 7;5(50):eaba7350. doi: 10.1126/sciimmunol.aba7350. Sci Immunol. 2020. PMID: 32769172 Free PMC article.
-
Odd-paired is a pioneer-like factor that coordinates with Zelda to control gene expression in embryos.Elife. 2020 Jul 23;9:e59610. doi: 10.7554/eLife.59610. Elife. 2020. PMID: 32701060 Free PMC article.
-
Single-cell multiome of the human retina and deep learning nominate causal variants in complex eye diseases.Cell Genom. 2022 Aug 10;2(8):100164. doi: 10.1016/j.xgen.2022.100164. Epub 2022 Jul 27. Cell Genom. 2022. PMID: 36277849 Free PMC article.
-
Negative elongation factor regulates muscle progenitor expansion for efficient myofiber repair and stem cell pool repopulation.Dev Cell. 2021 Apr 5;56(7):1014-1029.e7. doi: 10.1016/j.devcel.2021.02.025. Epub 2021 Mar 17. Dev Cell. 2021. PMID: 33735618 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
- U24HG009446/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- U24 HG009446/HG/NHGRI NIH HHS/United States
- U41 HG007000/HG/NHGRI NIH HHS/United States
- U41 HG009293/HG/NHGRI NIH HHS/United States
- T32 GM007315/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
