

Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys
- PMID: 31066324
- PMCID: PMC6844514
- DOI: 10.1080/15548627.2019.1615822
Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys
Abstract
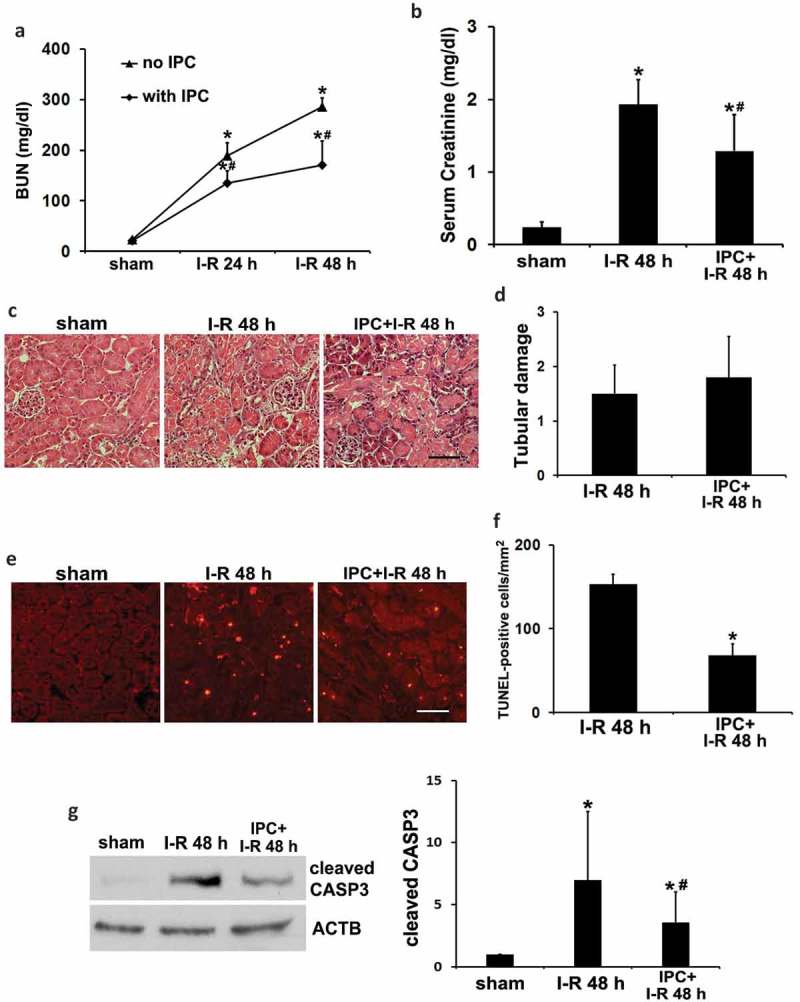
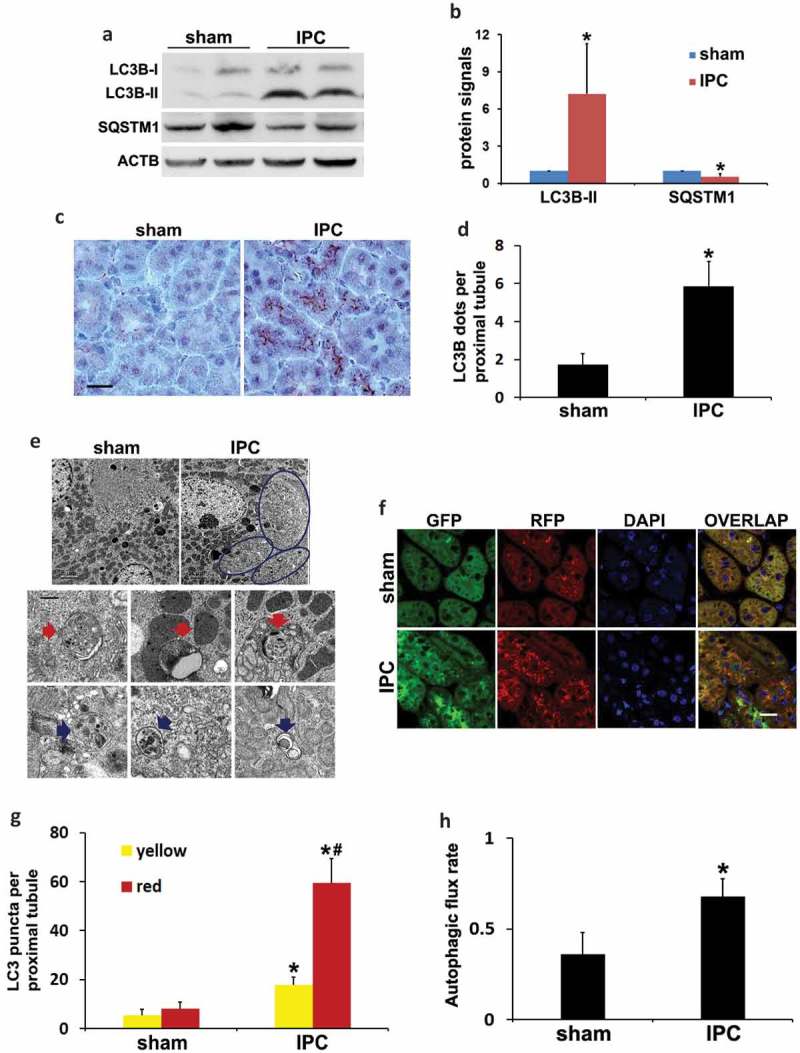
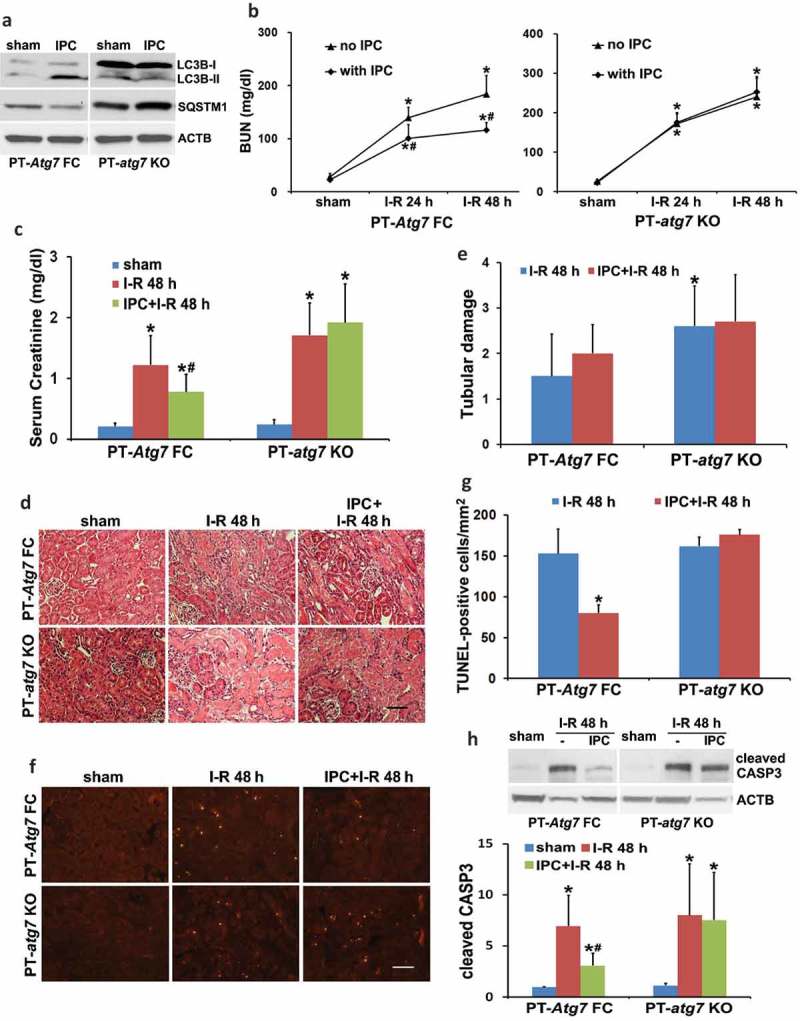
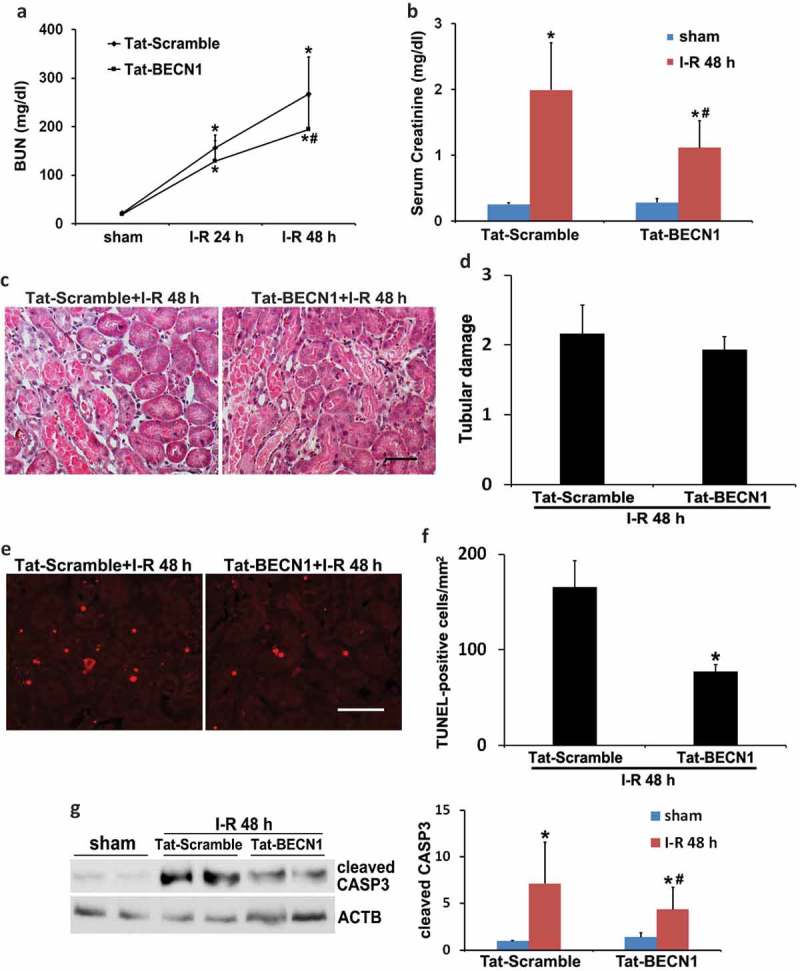
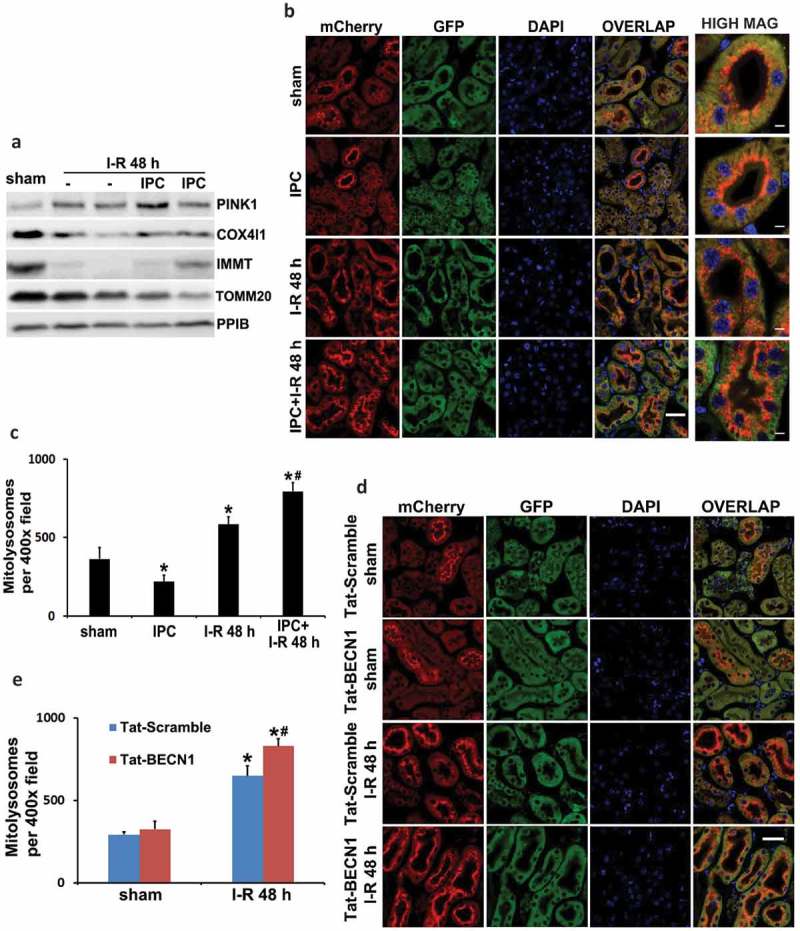
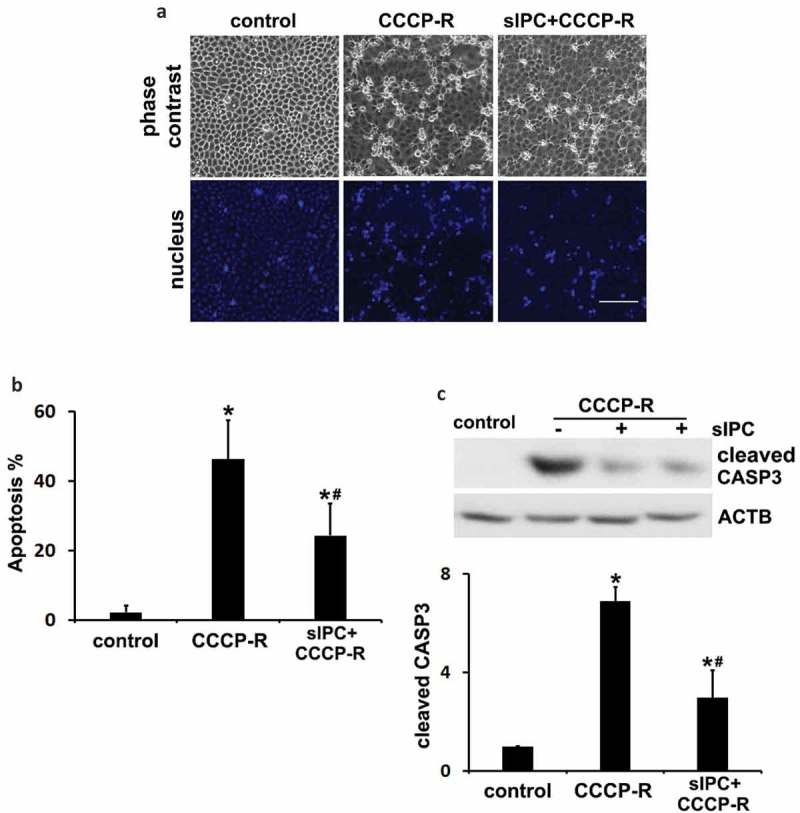
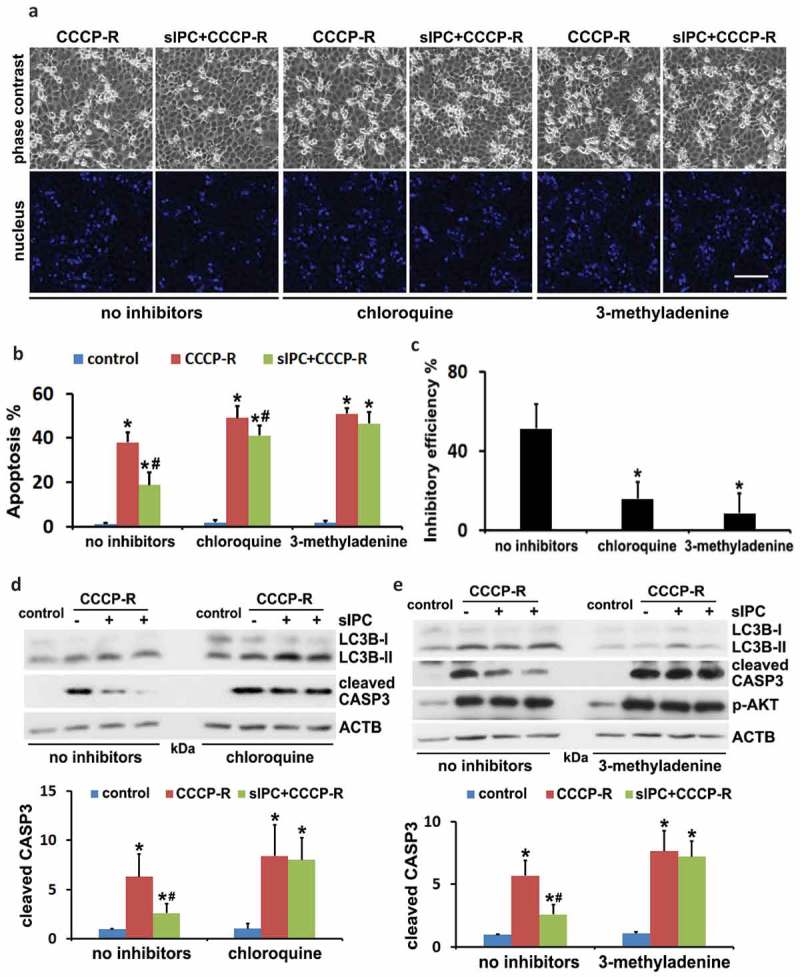
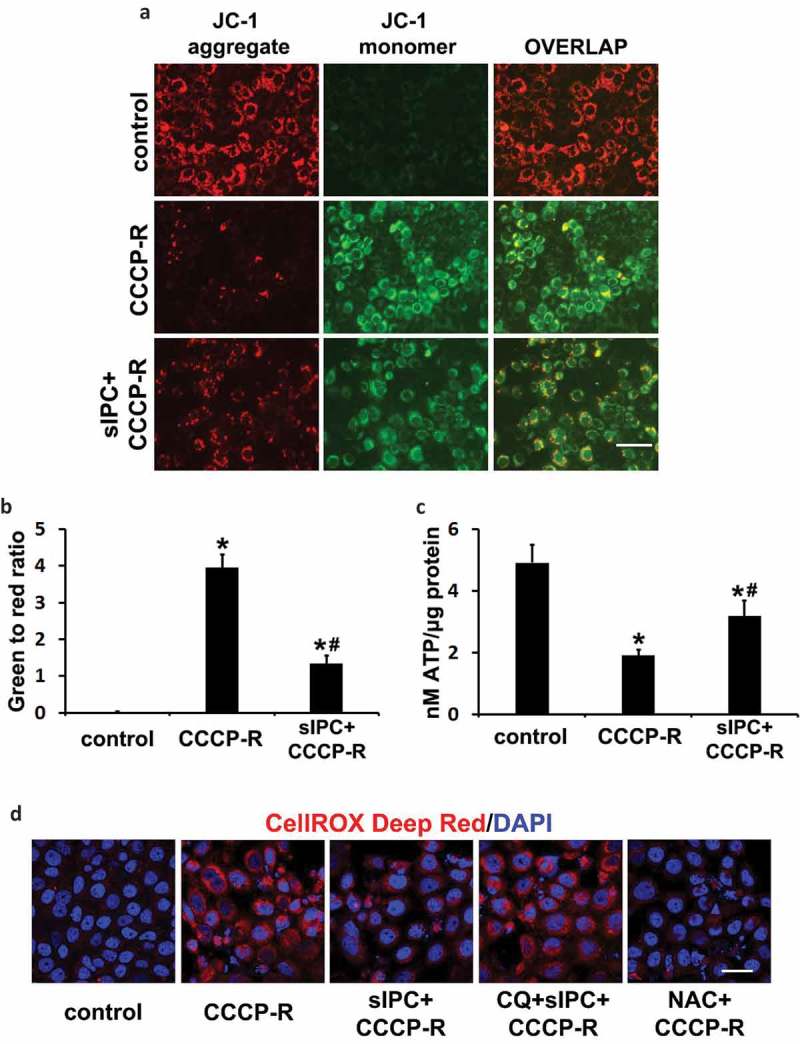
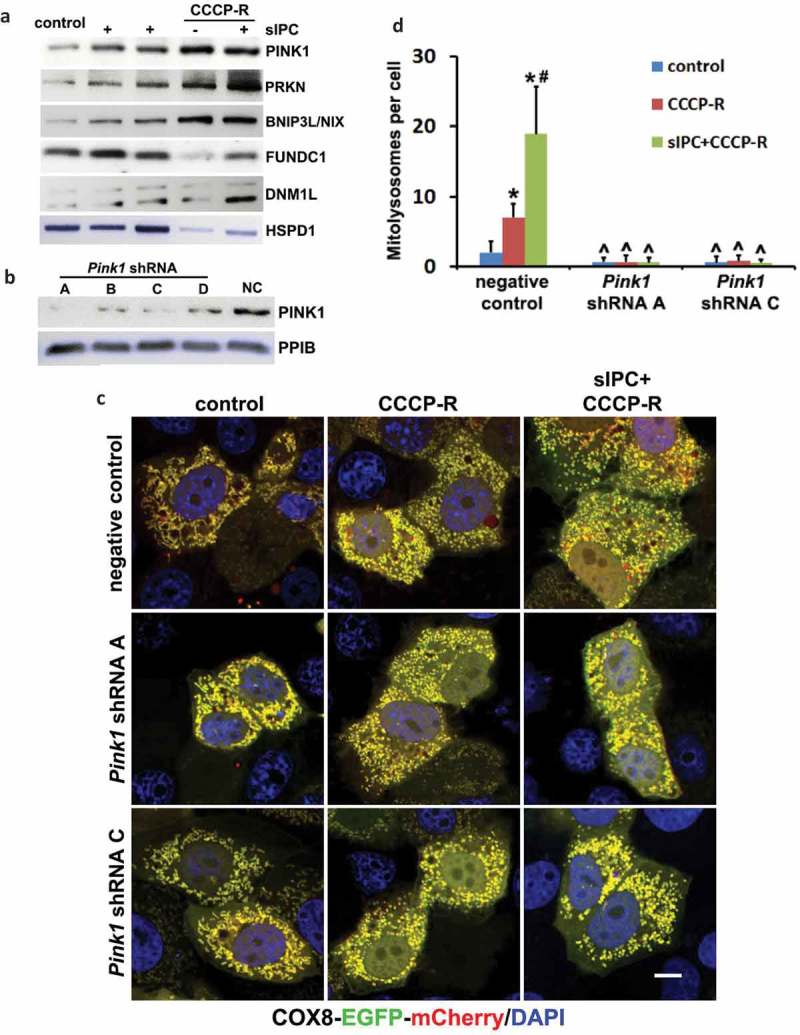
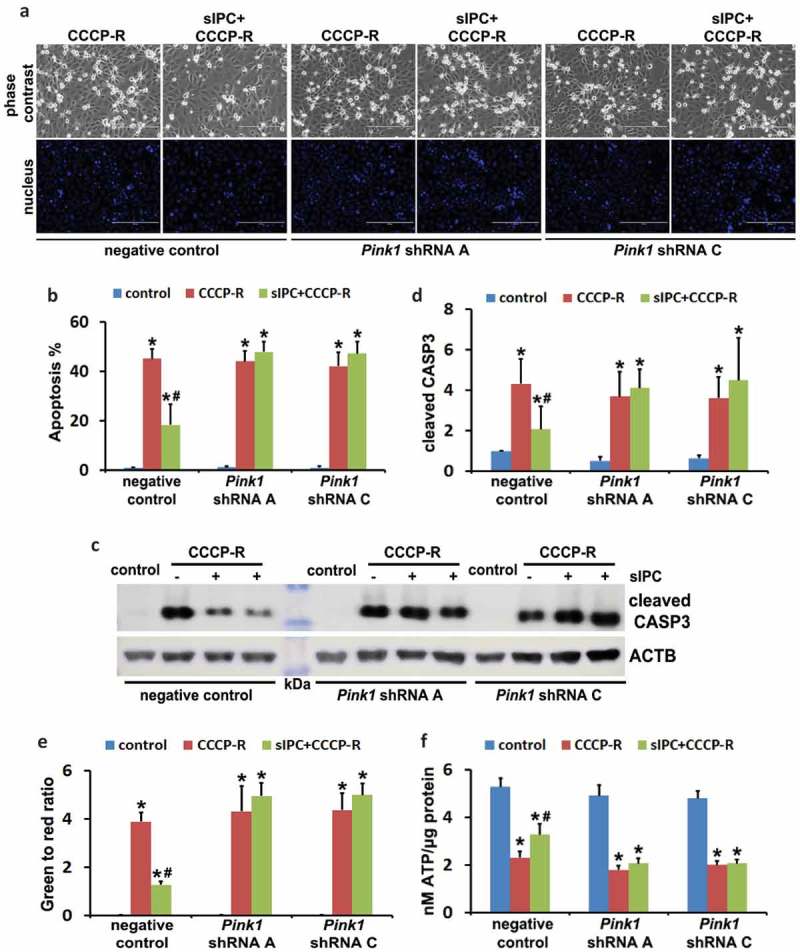
Ischemic preconditioning (IPC) affords tissue protection in organs including kidneys; however, the underlying mechanism remains unclear. Here we demonstrate an important role of macroautophagy/autophagy (especially mitophagy) in the protective effect of IPC in kidneys. IPC induced autophagy in renal tubular cells in mice and suppressed subsequent renal ischemia-reperfusion injury (IRI). The protective effect of IPC was abolished by pharmacological inhibitors of autophagy and by the ablation of Atg7 from kidney proximal tubules. Pretreatment with BECN1/Beclin1 peptide induced autophagy and protected against IRI. These results suggest the dependence of IPC protection on renal autophagy. During IPC, the mitophagy regulator PINK1 (PTEN induced putative kinase 1) was activated. Both IPC and BECN1 peptide enhanced mitolysosome formation during renal IRI in mitophagy reporter mice, suggesting that IPC may protect kidneys by activating mitophagy. We further established an in vitro model of IPC by inducing 'chemical ischemia' in kidney proximal tubular cells with carbonyl cyanide 3-chlorophenylhydrazone (CCCP). Brief treatment with CCCP protected against subsequent injury in these cells and the protective effect was abrogated by autophagy inhibition. In vitro IPC increased mitophagosome formation, enhanced the delivery of mitophagosomes to lysosomes, and promoted the clearance of damaged mitochondria during subsequent CCCP treatment. IPC also suppressed mitochondrial depolarization, improved ATP production, and inhibited the generation of reactive oxygen species. Knockdown of Pink1 suppressed mitophagy and reduced the cytoprotective effect of IPC. Together, these results suggest that autophagy, especially mitophagy, plays an important role in the protective effect of IPC.Abbreviations: ACTB: actin, beta; ATG: autophagy related; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; BUN: blood urea nitrogen; CASP3: caspase 3; CCCP: carbonyl cyanide 3-chlorophenylhydrazone; COX4I1: cytochrome c oxidase subunit 4I1; COX8: cytochrome c oxidase subunit 8; DAPI: 4',6-diamidino-2-phenylindole; DNM1L: dynamin 1 like; EGFP: enhanced green fluorescent protein; EM: electron microscopy; ER: endoplasmic reticulum; FC: floxed control; FIS1: fission, mitochondrial 1; FUNDC1: FUN14 domain containing 1; H-E: hematoxylin-eosin; HIF1A: hypoxia inducible factor 1 subunit alpha; HSPD1: heat shock protein family D (Hsp60) member 1; IMMT/MIC60: inner membrane mitochondrial protein; IPC: ischemic preconditioning; I-R: ischemia-reperfusion; IRI: ischemia-reperfusion injury; JC-1: 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide; KO: knockout; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; mito-QC: mito-quality control; mRFP: monomeric red fluorescent protein; NAC: N-acetylcysteine; PINK1: PTEN induced putative kinase 1; PPIB: peptidylprolyl isomerase B; PRKN: parkin RBR E3 ubiquitin protein ligase; ROS: reactive oxygen species; RPTC: rat proximal tubular cells; SD: standard deviation; sIPC: simulated IPC; SQSTM1/p62: sequestosome 1; TOMM20: translocase of outer mitochondrial membrane 20; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
Keywords: Acute kidney injury; autophagy; ischemic preconditioning; mitophagy; proximal tubule; renal ischemia-reperfusion.
Figures
Similar articles
-
Mitochondria ROS and mitophagy in acute kidney injury.Autophagy. 2023 Feb;19(2):401-414. doi: 10.1080/15548627.2022.2084862. Epub 2022 Jun 9. Autophagy. 2023. PMID: 35678504 Free PMC article.
-
BNIP3L-mediated mitophagy is required for mitochondrial remodeling during the differentiation of optic nerve oligodendrocytes.Autophagy. 2021 Oct;17(10):3140-3159. doi: 10.1080/15548627.2020.1871204. Epub 2021 Jan 19. Autophagy. 2021. PMID: 33404293 Free PMC article.
-
BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains.Autophagy. 2021 Aug;17(8):1934-1946. doi: 10.1080/15548627.2020.1802089. Epub 2020 Aug 12. Autophagy. 2021. PMID: 32722981 Free PMC article.
-
Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles.Autophagy. 2021 Feb;17(2):385-401. doi: 10.1080/15548627.2020.1725377. Epub 2020 Feb 12. Autophagy. 2021. PMID: 32048886 Free PMC article. Review.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
Cited by
-
Cellular senescence of renal tubular epithelial cells in acute kidney injury.Cell Death Discov. 2024 Feb 5;10(1):62. doi: 10.1038/s41420-024-01831-9. Cell Death Discov. 2024. PMID: 38316761 Free PMC article. Review.
-
Mitophagy-A New Target of Bone Disease.Biomolecules. 2022 Oct 4;12(10):1420. doi: 10.3390/biom12101420. Biomolecules. 2022. PMID: 36291629 Free PMC article. Review.
-
Cyclovirobuxine D Induced-Mitophagy through the p65/BNIP3/LC3 Axis Potentiates Its Apoptosis-Inducing Effects in Lung Cancer Cells.Int J Mol Sci. 2021 May 29;22(11):5820. doi: 10.3390/ijms22115820. Int J Mol Sci. 2021. PMID: 34072333 Free PMC article.
-
Ginsenoside Rg3-loaded, reactive oxygen species-responsive polymeric nanoparticles for alleviating myocardial ischemia-reperfusion injury.J Control Release. 2020 Jan 10;317:259-272. doi: 10.1016/j.jconrel.2019.11.032. Epub 2019 Nov 26. J Control Release. 2020. PMID: 31783047 Free PMC article.
-
METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy.J Exp Clin Cancer Res. 2023 Mar 17;42(1):65. doi: 10.1186/s13046-023-02638-9. J Exp Clin Cancer Res. 2023. PMID: 36932427 Free PMC article.
References
-
- Yellon DM, Downey JM.. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003. October;83(4):1113–1151. . PubMed PMID: 14506302. - PubMed
-
- Pasupathy S, Homer-Vanniasinkam S. Ischaemic preconditioning protects against ischaemia/reperfusion injury: emerging concepts. Eur J Vasc Endovasc Surg. 2005. February;29(2):106–115. . PubMed PMID: 15649715. - PubMed
-
- Bonventre JV. Kidney ischemic preconditioning. Curr Opin Nephrol Hypertens. 2002. January;11(1):43–48. PubMed PMID: 11753086. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous