

TOP2β-Dependent Nuclear DNA Damage Shapes Extracellular Growth Factor Responses via Dynamic AKT Phosphorylation to Control Virus Latency
- PMID: 30930055
- PMCID: PMC6499694
- DOI: 10.1016/j.molcel.2019.02.032
TOP2β-Dependent Nuclear DNA Damage Shapes Extracellular Growth Factor Responses via Dynamic AKT Phosphorylation to Control Virus Latency
Abstract
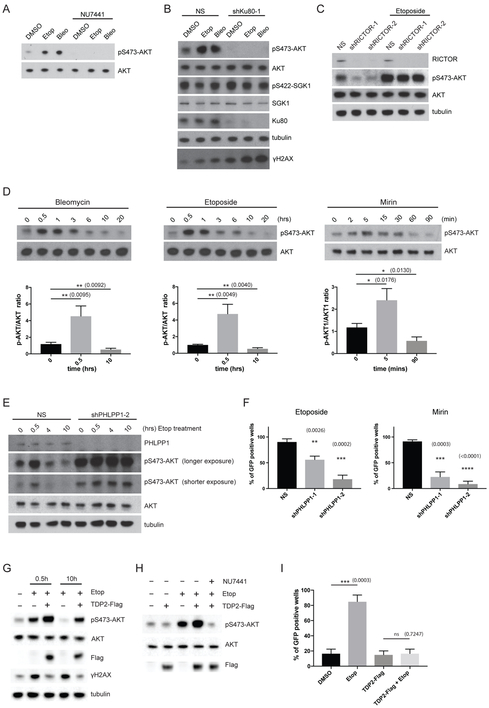
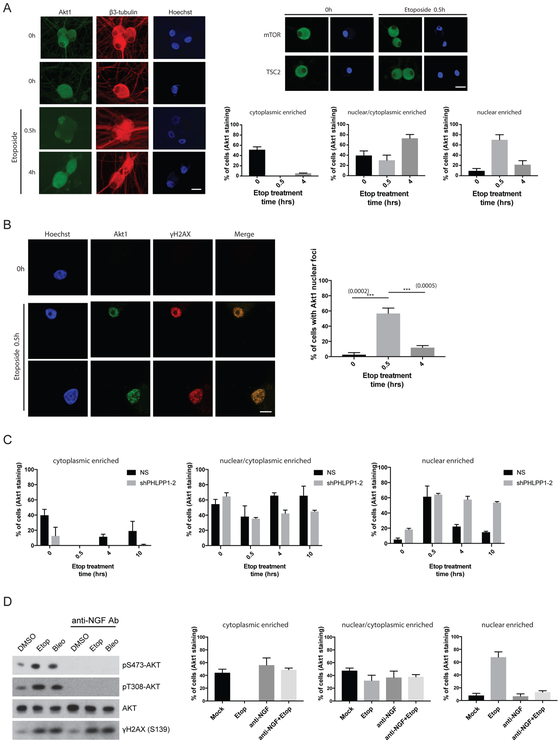
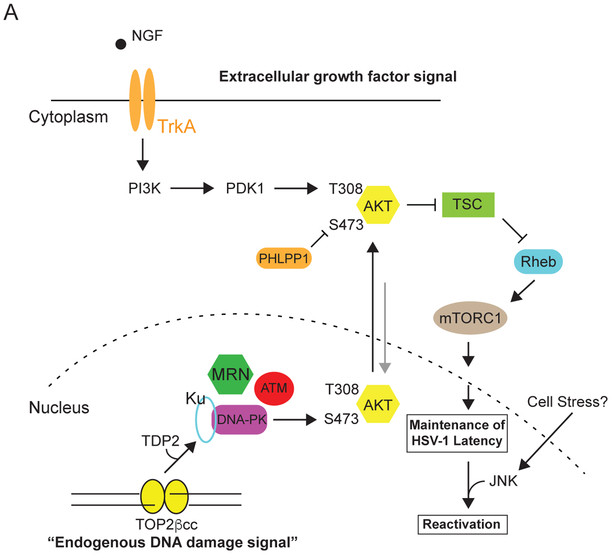
The mTOR pathway integrates both extracellular and intracellular signals and serves as a central regulator of cell metabolism, growth, survival, and stress responses. Neurotropic viruses, such as herpes simplex virus-1 (HSV-1), also rely on cellular AKT-mTORC1 signaling to achieve viral latency. Here, we define a novel genotoxic response whereby spatially separated signals initiated by extracellular neurotrophic factors and nuclear DNA damage are integrated by the AKT-mTORC1 pathway. We demonstrate that endogenous DNA double-strand breaks (DSBs) mediated by Topoisomerase 2β-DNA cleavage complex (TOP2βcc) intermediates are required to achieve AKT-mTORC1 signaling and maintain HSV-1 latency in neurons. Suppression of host DNA-repair pathways that remove TOP2βcc trigger HSV-1 reactivation. Moreover, perturbation of AKT phosphorylation dynamics by downregulating the PHLPP1 phosphatase led to AKT mis-localization and disruption of DSB-induced HSV-1 reactivation. Thus, the cellular genome integrity and environmental inputs are consolidated and co-opted by a latent virus to balance lifelong infection with transmission.
Keywords: AKT; DNA-PK; Herpes simplex virus-1 (HSV-1); Mre11-Rad50-Nbs1 (MRN); Non-homologous end-joining (NHEJ); PHLPP1; mTORC1; topoisomerase 2 beta (TOP2b); tyrosyl-DNA-phosphodiesterase 2 (TDP2); viral latency.
Copyright © 2019 Elsevier Inc. All rights reserved.
Figures
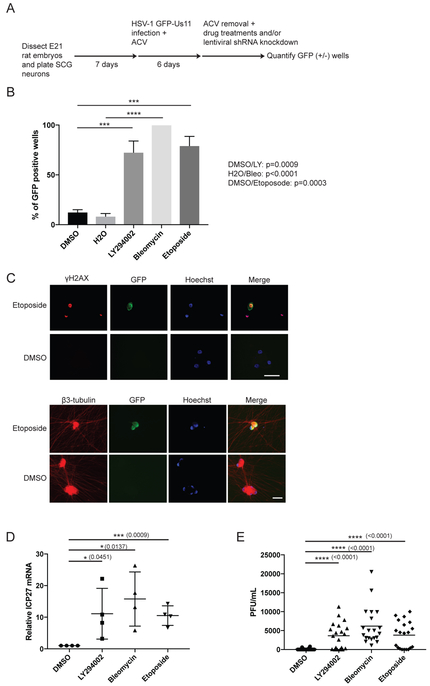
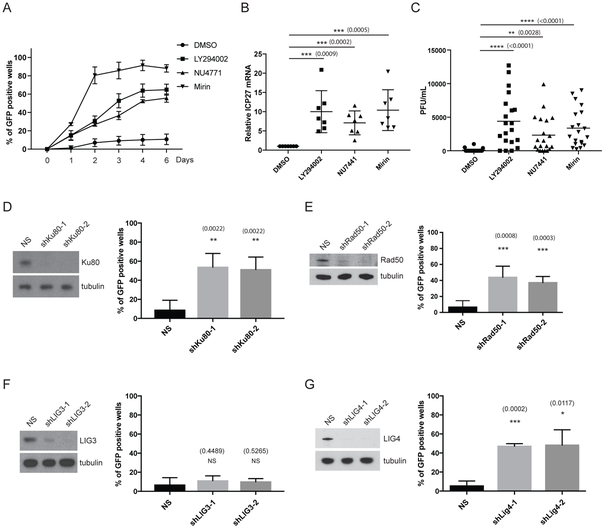
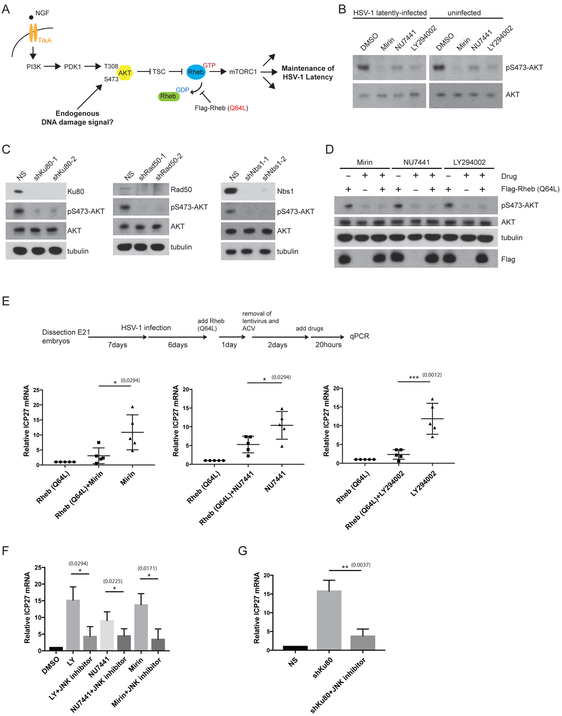
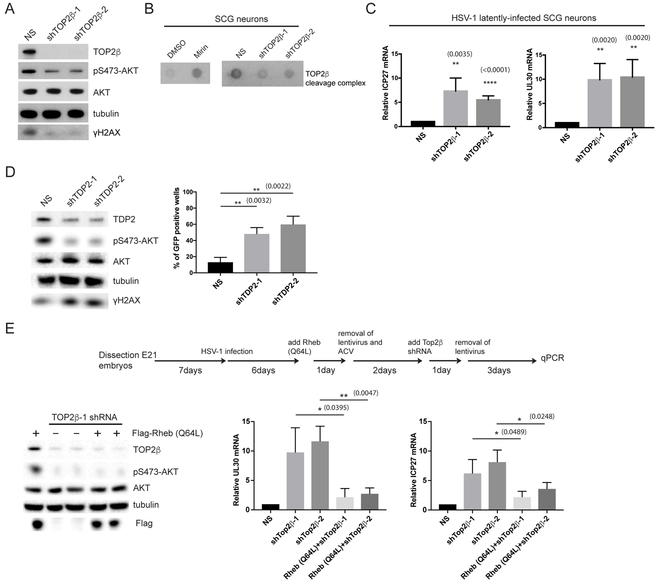
Comment in
-
DNA Damage Meets Neurotrophin Signaling: A Delicate Balancing AKT to Maintain Virus Latency.Mol Cell. 2019 May 2;74(3):411-413. doi: 10.1016/j.molcel.2019.04.015. Mol Cell. 2019. PMID: 31051136
Similar articles
-
Impact of the MRN Complex on Adeno-Associated Virus Integration and Replication during Coinfection with Herpes Simplex Virus 1.J Virol. 2015 Jul;89(13):6824-34. doi: 10.1128/JVI.00171-15. Epub 2015 Apr 22. J Virol. 2015. PMID: 25903339 Free PMC article.
-
Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA.Genes (Basel). 2019 Oct 30;10(11):868. doi: 10.3390/genes10110868. Genes (Basel). 2019. PMID: 31671674 Free PMC article. Review.
-
AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11.Oncogene. 2018 Jan 25;37(4):427-438. doi: 10.1038/onc.2017.340. Epub 2017 Oct 2. Oncogene. 2018. PMID: 28967905 Free PMC article.
-
Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes.Mol Cell. 2016 Nov 3;64(3):580-592. doi: 10.1016/j.molcel.2016.10.011. Mol Cell. 2016. PMID: 27814490
-
Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair.Prog Biophys Mol Biol. 2015 Mar;117(2-3):182-193. doi: 10.1016/j.pbiomolbio.2014.12.004. Epub 2015 Jan 7. Prog Biophys Mol Biol. 2015. PMID: 25576492 Free PMC article. Review.
Cited by
-
DNA methylation regulates RNA m6A modification through transcription factor SP1 during the development of porcine somatic cell nuclear transfer embryos.Cell Prolif. 2024 May;57(5):e13581. doi: 10.1111/cpr.13581. Epub 2023 Dec 14. Cell Prolif. 2024. PMID: 38095020 Free PMC article.
-
Ex Vivo Herpes Simplex Virus Reactivation Involves a Dual Leucine Zipper Kinase-Dependent Wave of Lytic Gene Expression That Is Independent of Histone Demethylase Activity and Viral Genome Synthesis.J Virol. 2022 Jun 22;96(12):e0047522. doi: 10.1128/jvi.00475-22. Epub 2022 May 23. J Virol. 2022. PMID: 35604215 Free PMC article.
-
Herpes Simplex Virus 1 Manipulates Host Cell Antiviral and Proviral DNA Damage Responses.mBio. 2021 Feb 9;12(1):e03552-20. doi: 10.1128/mBio.03552-20. mBio. 2021. PMID: 33563816 Free PMC article.
-
Topoisomerase 2β Induces DNA Breaks To Regulate Human Papillomavirus Replication.mBio. 2021 Feb 9;12(1):e00005-21. doi: 10.1128/mBio.00005-21. mBio. 2021. PMID: 33563836 Free PMC article.
-
Deletion of the Transcriptional Coactivator HCF-1 In Vivo Impairs the Removal of Repressive Heterochromatin from Latent HSV Genomes and Suppresses the Initiation of Viral Reactivation.mBio. 2023 Feb 28;14(1):e0354222. doi: 10.1128/mbio.03542-22. Epub 2023 Jan 24. mBio. 2023. PMID: 36692302 Free PMC article.
References
-
- Bencherit D, Remy S, Le Vern Y, Vychodil T, Bertzbach LD, Kaufer BB, Denesvre C, and Trapp-Fragnet L (2017). Induction of DNA Damages upon Marek's Disease Virus Infection: Implication in Viral Replication and Pathogenesis. J Virol 91, pii: e01658–01617. doi: 01610.01128/JVI.01658-01617. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
