

Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension
- PMID: 30929899
- PMCID: PMC6610881
- DOI: 10.1016/j.cell.2019.02.013
Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension
Abstract
Autophagy is required in diverse paradigms of lifespan extension, leading to the prevailing notion that autophagy is beneficial for longevity. However, why autophagy is harmful in certain contexts remains unexplained. Here, we show that mitochondrial permeability defines the impact of autophagy on aging. Elevated autophagy unexpectedly shortens lifespan in C. elegans lacking serum/glucocorticoid regulated kinase-1 (sgk-1) because of increased mitochondrial permeability. In sgk-1 mutants, reducing levels of autophagy or mitochondrial permeability transition pore (mPTP) opening restores normal lifespan. Remarkably, low mitochondrial permeability is required across all paradigms examined of autophagy-dependent lifespan extension. Genetically induced mPTP opening blocks autophagy-dependent lifespan extension resulting from caloric restriction or loss of germline stem cells. Mitochondrial permeability similarly transforms autophagy into a destructive force in mammals, as liver-specific Sgk knockout mice demonstrate marked enhancement of hepatocyte autophagy, mPTP opening, and death with ischemia/reperfusion injury. Targeting mitochondrial permeability may maximize benefits of autophagy in aging.
Keywords: SGK; aging; autophagy; ischemia/reperfusion injury; longevity; mPTP; mTORC2; mitochondrial permeability.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
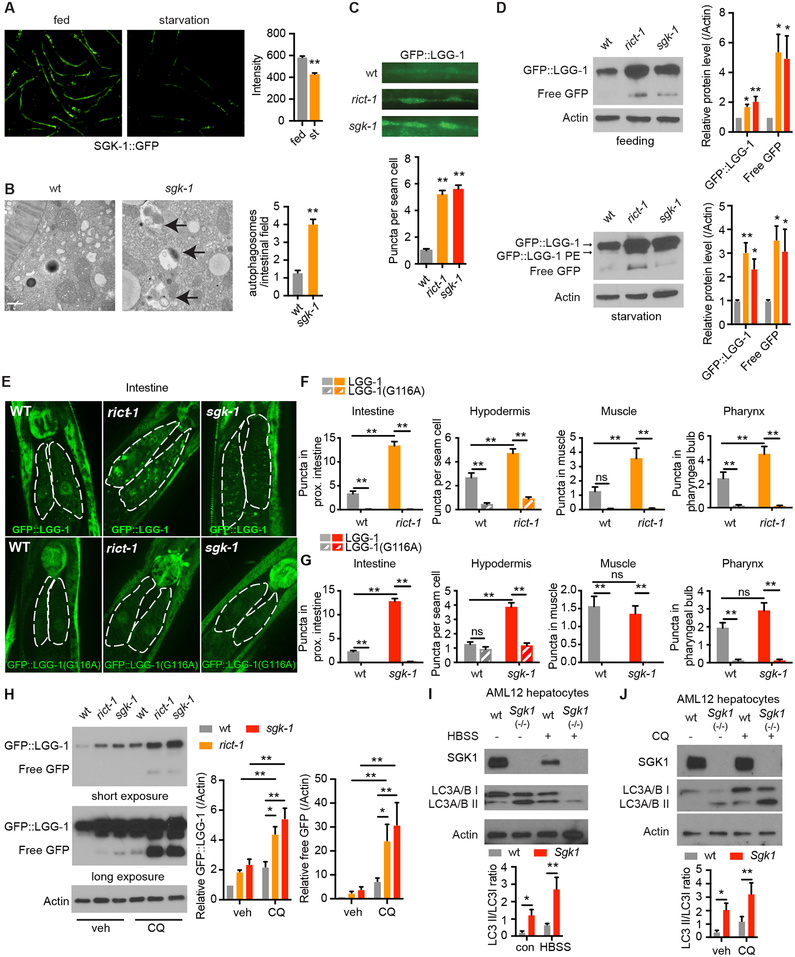
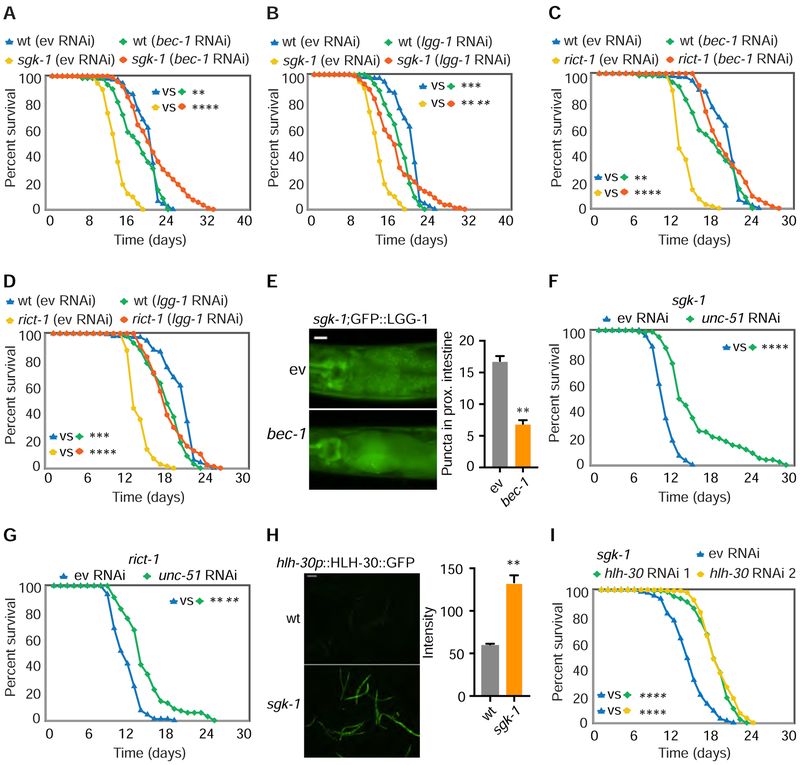
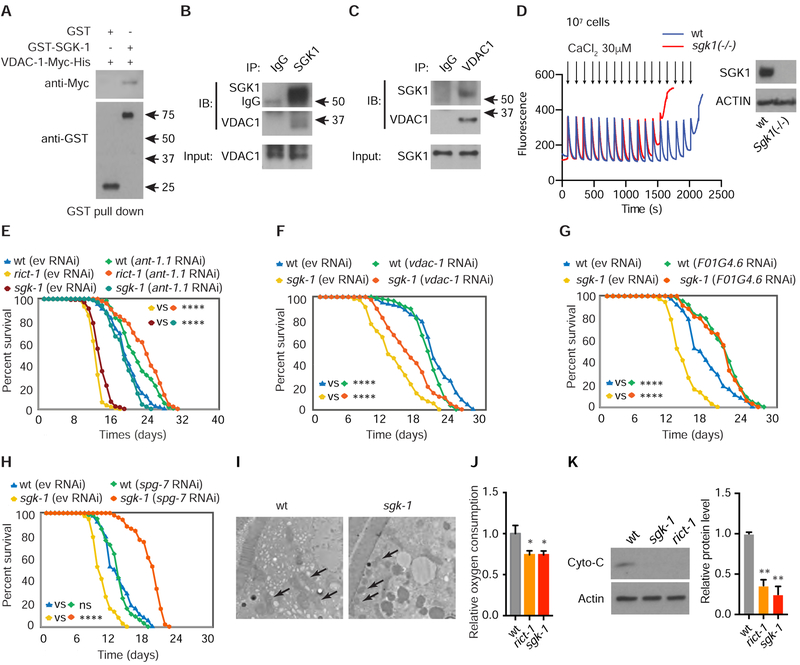
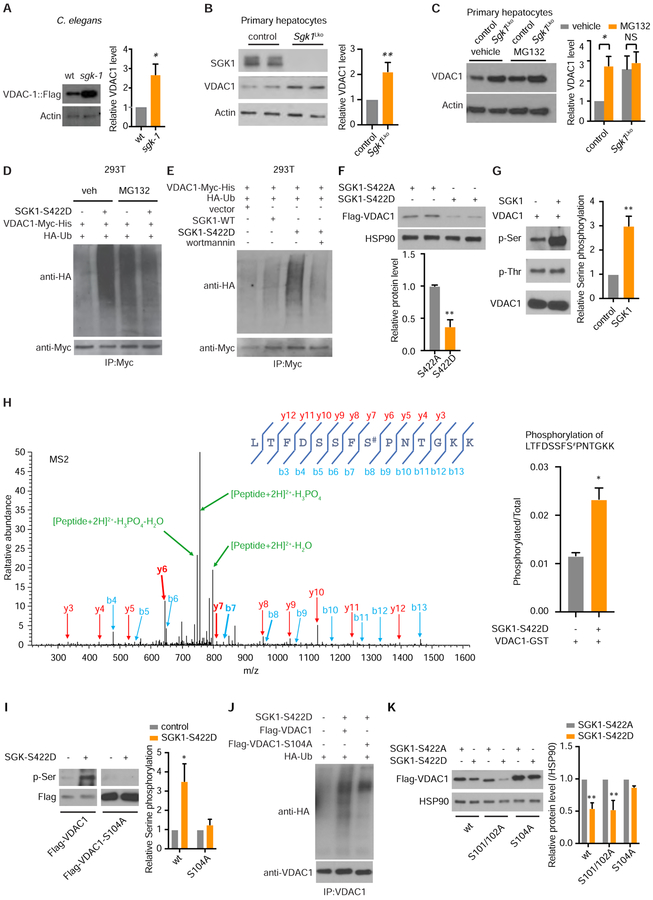
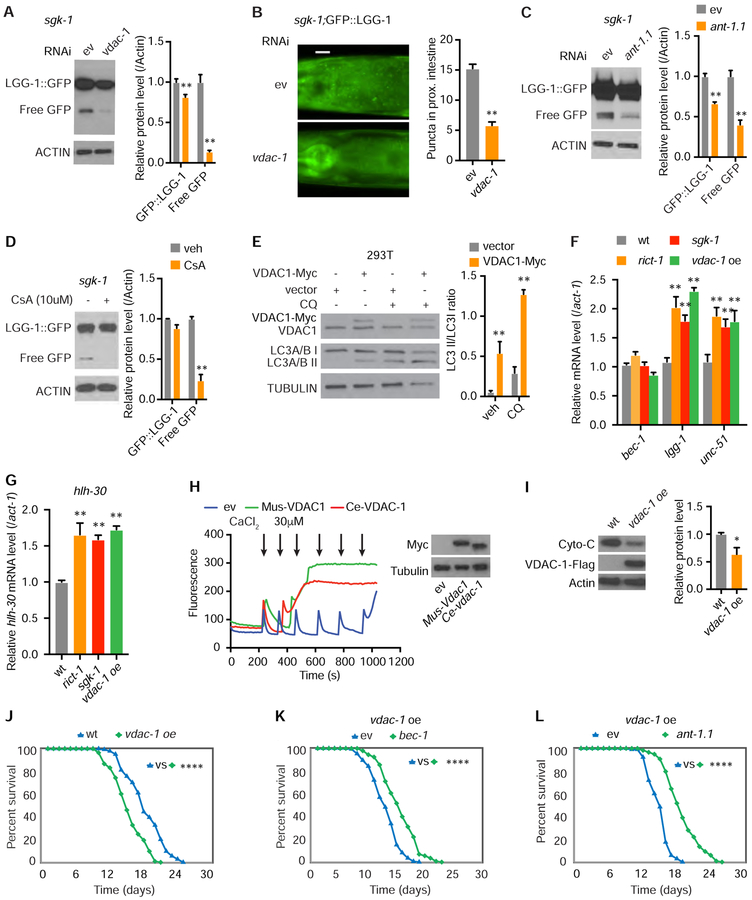
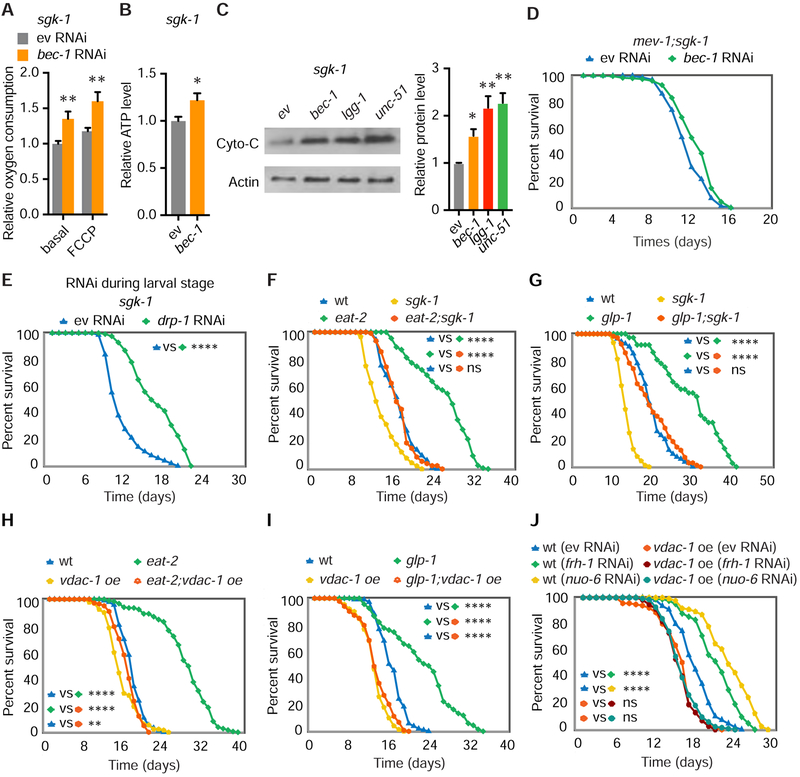
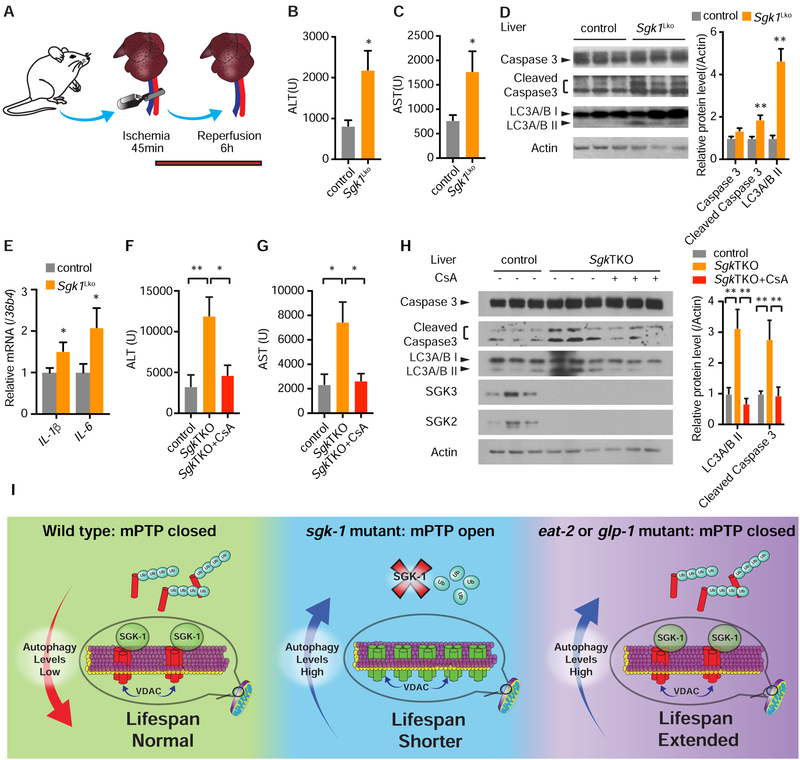
Comment in
-
Does Autophagy Promote Longevity? It Depends.Cell. 2019 Apr 4;177(2):221-222. doi: 10.1016/j.cell.2019.03.021. Cell. 2019. PMID: 30951663
-
Autophagy promotes longevity-except in the presence of 'leaky' mitochondria.Cardiovasc Res. 2019 Oct 1;115(12):e118-e120. doi: 10.1093/cvr/cvz224. Cardiovasc Res. 2019. PMID: 31544942 No abstract available.
Similar articles
-
Suppressing the dark side of autophagy.Autophagy. 2019 Oct;15(10):1852-1853. doi: 10.1080/15548627.2019.1644077. Epub 2019 Jul 18. Autophagy. 2019. PMID: 31313620 Free PMC article.
-
Prohibitin depletion extends lifespan of a TORC2/SGK-1 mutant through autophagy and the mitochondrial UPR.Aging Cell. 2021 May;20(5):e13359. doi: 10.1111/acel.13359. Epub 2021 May 3. Aging Cell. 2021. PMID: 33939875 Free PMC article.
-
Prohibitin-mediated lifespan and mitochondrial stress implicate SGK-1, insulin/IGF and mTORC2 in C. elegans.PLoS One. 2014 Sep 29;9(9):e107671. doi: 10.1371/journal.pone.0107671. eCollection 2014. PLoS One. 2014. PMID: 25265021 Free PMC article.
-
The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore.Aging Cell. 2017 Oct;16(5):943-955. doi: 10.1111/acel.12650. Epub 2017 Jul 31. Aging Cell. 2017. PMID: 28758328 Free PMC article. Review.
-
The Mitochondrial Permeability Transition: Nexus of Aging, Disease and Longevity.Cells. 2021 Jan 6;10(1):79. doi: 10.3390/cells10010079. Cells. 2021. PMID: 33418876 Free PMC article. Review.
Cited by
-
Plasma methionine metabolic profile is associated with longevity in mammals.Commun Biol. 2021 Jun 11;4(1):725. doi: 10.1038/s42003-021-02254-3. Commun Biol. 2021. PMID: 34117367 Free PMC article.
-
Macroautophagy and aging: The impact of cellular recycling on health and longevity.Mol Aspects Med. 2021 Dec;82:101020. doi: 10.1016/j.mam.2021.101020. Epub 2021 Sep 7. Mol Aspects Med. 2021. PMID: 34507801 Free PMC article. Review.
-
SGK-1 mediated inhibition of iron import is a determinant of lifespan in C. elegans.MicroPubl Biol. 2023 Sep 17;2023:10.17912/micropub.biology.000970. doi: 10.17912/micropub.biology.000970. eCollection 2023. MicroPubl Biol. 2023. PMID: 37799207 Free PMC article.
-
Dietary serine-microbiota interaction enhances chemotherapeutic toxicity without altering drug conversion.Nat Commun. 2020 May 22;11(1):2587. doi: 10.1038/s41467-020-16220-w. Nat Commun. 2020. PMID: 32444616 Free PMC article.
-
Icariin Ameliorates D-galactose-induced Cell Injury in Neuron-like PC12 Cells by Inhibiting MPTP Opening.Curr Med Sci. 2024 Aug;44(4):748-758. doi: 10.1007/s11596-024-2892-0. Epub 2024 Jun 20. Curr Med Sci. 2024. PMID: 38900385
References
-
- Batandier C, Leverve X, and Fontaine E (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. The Journal of biological chemistry 279, 17197–17204. - PubMed
-
- Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, and Yellon DM (2008). Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol 103, 274–284. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
