

Stapled Peptides Inhibitors: A New Window for Target Drug Discovery
- PMID: 30867891
- PMCID: PMC6396041
- DOI: 10.1016/j.csbj.2019.01.012
Stapled Peptides Inhibitors: A New Window for Target Drug Discovery
Abstract
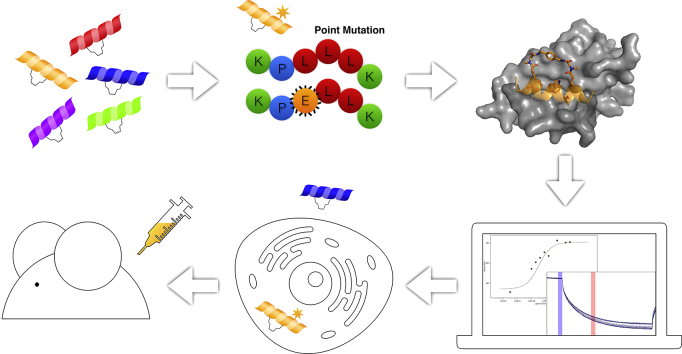
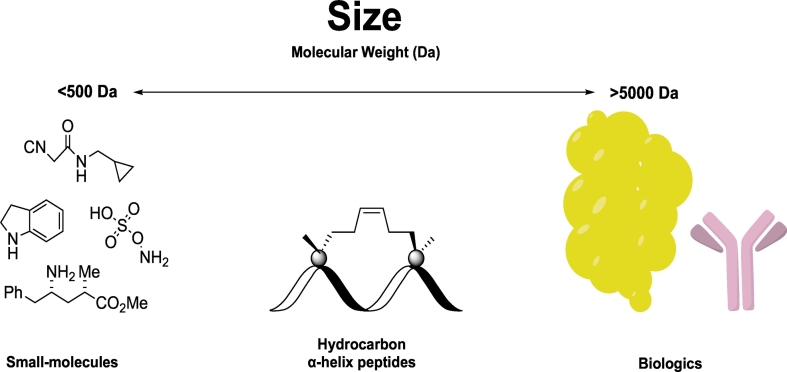
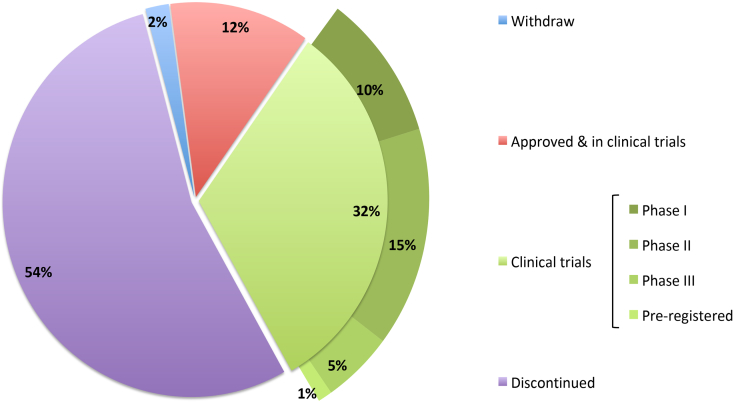
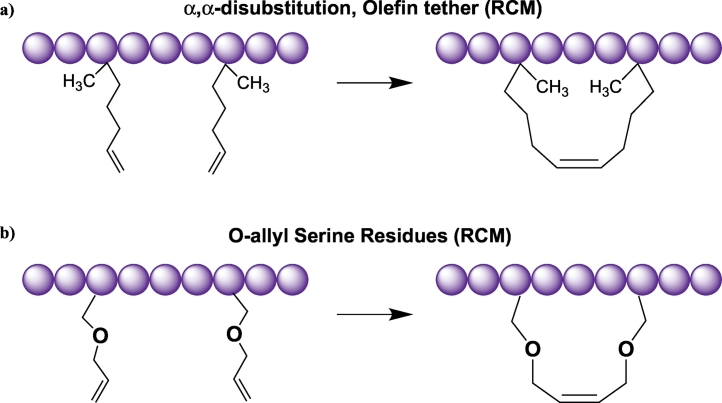
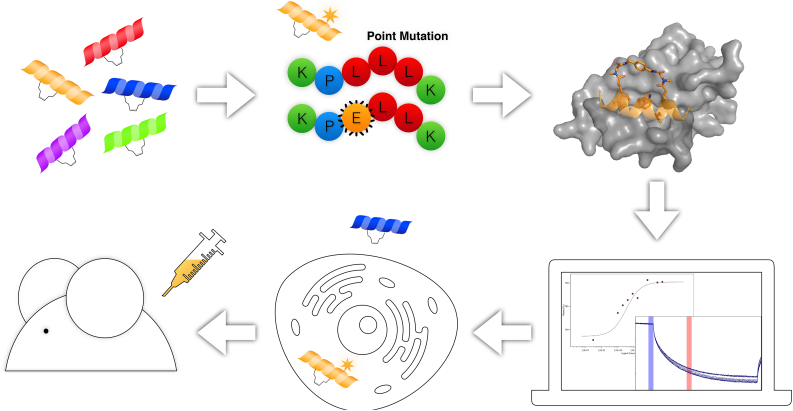
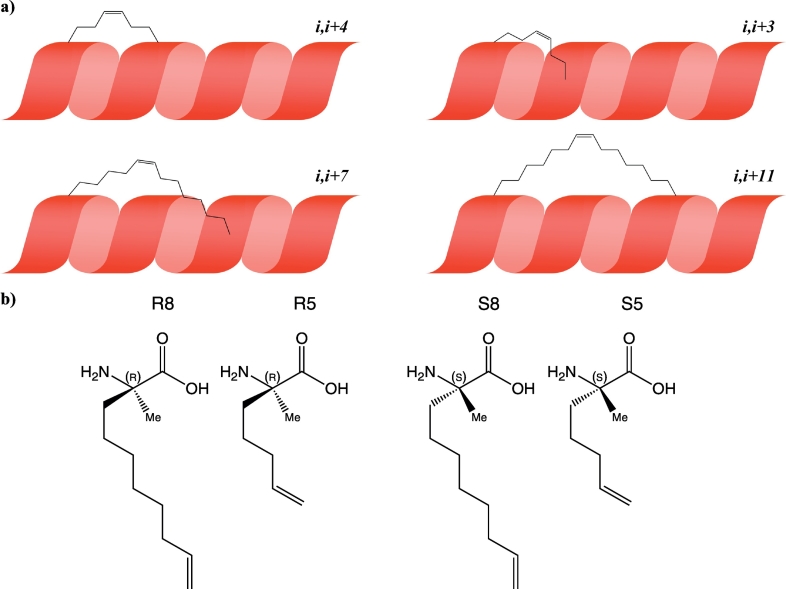
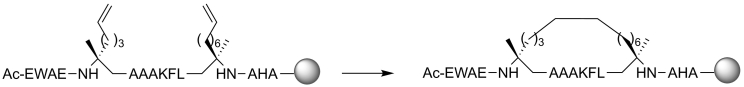
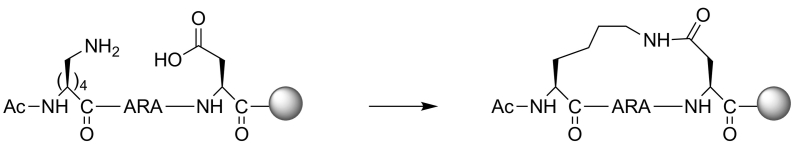
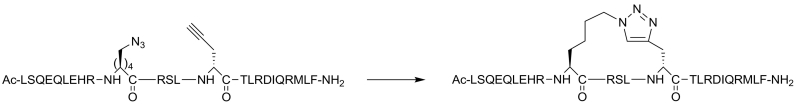
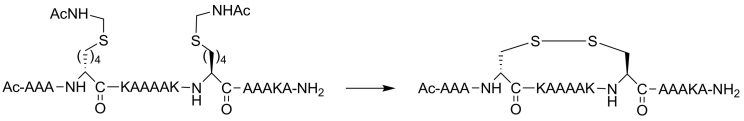
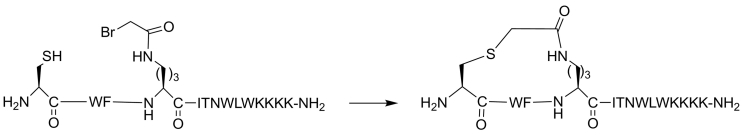
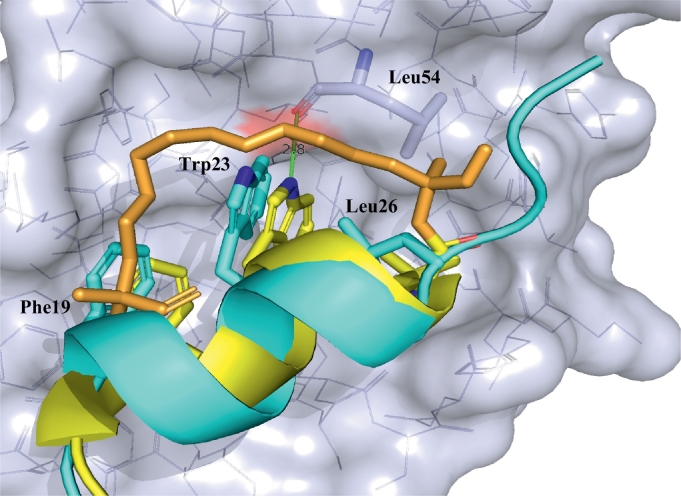
Protein-protein interaction (PPI) is a hot topic in clinical research as protein networking has a major impact in human disease. Such PPIs are potential drugs targets, leading to the need to inhibit/block specific PPIs. While small molecule inhibitors have had some success and reached clinical trials, they have generally failed to address the flat and large nature of PPI surfaces. As a result, larger biologics were developed for PPI surfaces and they have successfully targeted PPIs located outside the cell. However, biologics have low bioavailability and cannot reach intracellular targets. A novel class -hydrocarbon-stapled α-helical peptides that are synthetic mini-proteins locked into their bioactive structure through site-specific introduction of a chemical linker- has shown promise. Stapled peptides show an ability to inhibit intracellular PPIs that previously have been intractable with traditional small molecule or biologics, suggesting that they offer a novel therapeutic modality. In this review, we highlight what stapling adds to natural-mimicking peptides, describe the revolution of synthetic chemistry techniques and how current drug discovery approaches have been adapted to stabilize active peptide conformations, including ring-closing metathesis (RCM), lactamisation, cycloadditions and reversible reactions. We provide an overview on the available stapled peptide high-resolution structures in the protein data bank, with four selected structures discussed in details due to remarkable interactions of their staple with the target surface. We believe that stapled peptides are promising drug candidates and open the doors for peptide therapeutics to reach currently "undruggable" space.
Keywords: Drug discovery; Inhibitor; PPI; Stapled peptide; Synthetic chemistry.
Figures
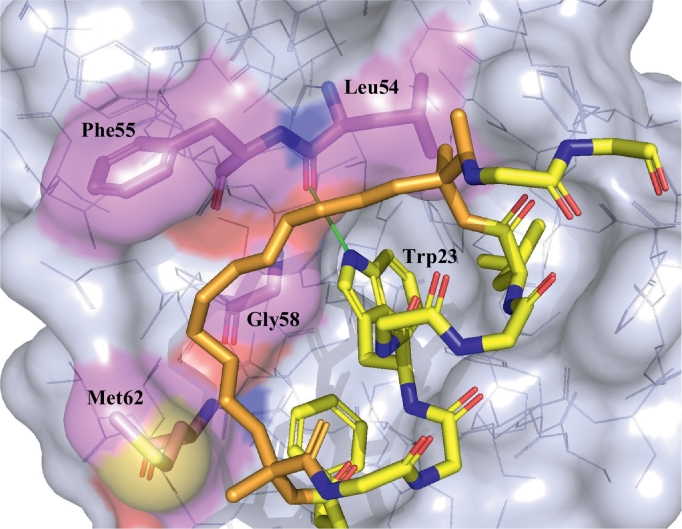
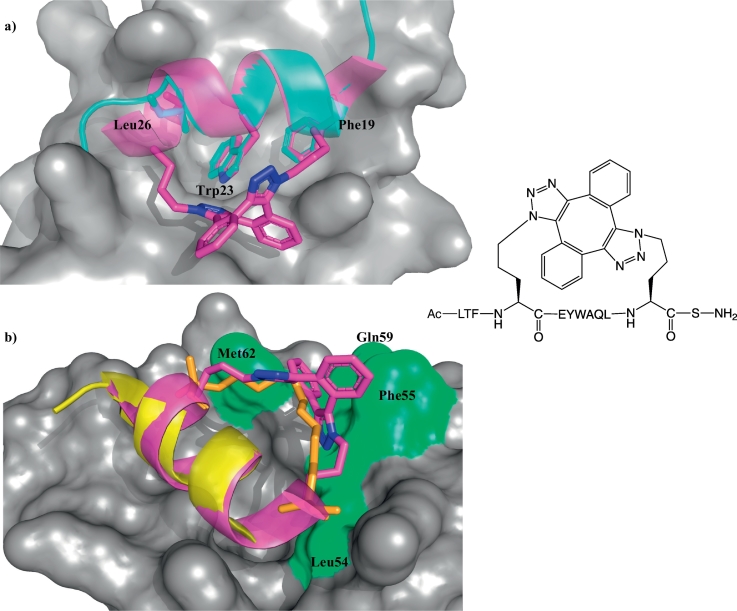
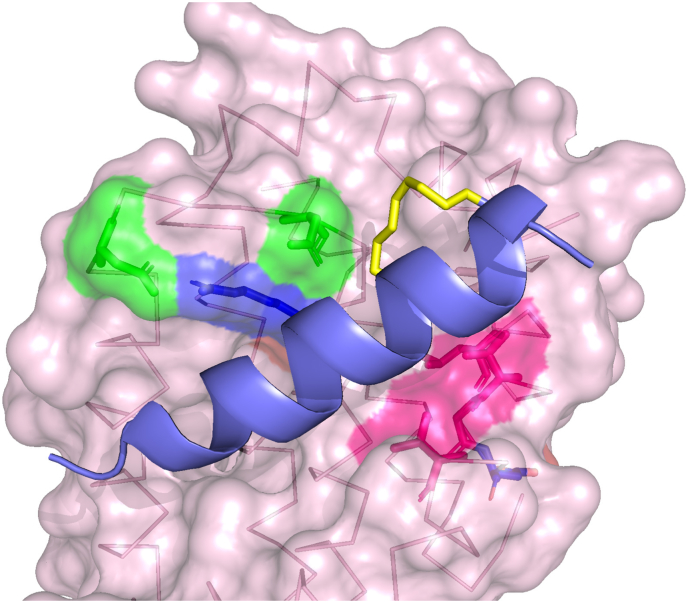
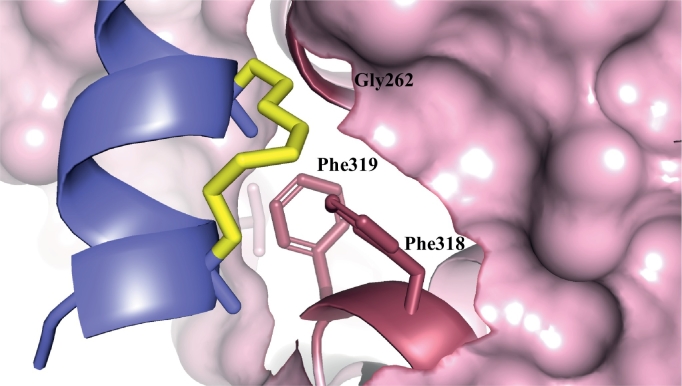
Similar articles
-
Peptide therapeutics: targeting the undruggable space.Eur J Med Chem. 2015 Apr 13;94:459-70. doi: 10.1016/j.ejmech.2015.01.014. Epub 2015 Jan 9. Eur J Med Chem. 2015. PMID: 25591543 Review.
-
Stapled peptides for intracellular drug targets.Methods Enzymol. 2012;503:3-33. doi: 10.1016/B978-0-12-396962-0.00001-X. Methods Enzymol. 2012. PMID: 22230563
-
Helical Stabilization of Peptide Macrocycles by Stapled Architectures.Methods Mol Biol. 2022;2371:391-409. doi: 10.1007/978-1-0716-1689-5_21. Methods Mol Biol. 2022. PMID: 34596860
-
Cyclobutane-bearing restricted anchoring residues enabled geometry-specific hydrocarbon peptide stapling.Chem Sci. 2023 Sep 29;14(41):11499-11506. doi: 10.1039/d3sc04279k. eCollection 2023 Oct 25. Chem Sci. 2023. PMID: 37886087 Free PMC article.
-
Stabilized cyclic peptides as modulators of protein-protein interactions: promising strategies and biological evaluation.RSC Med Chem. 2023 Oct 20;14(12):2496-2508. doi: 10.1039/d3md00487b. eCollection 2023 Dec 13. RSC Med Chem. 2023. PMID: 38107173 Free PMC article. Review.
Cited by
-
Discovery and Characterization of Peptide Inhibitors for Calcium and Integrin Binding Protein 1.ACS Chem Biol. 2020 Jun 19;15(6):1505-1516. doi: 10.1021/acschembio.0c00144. Epub 2020 May 26. ACS Chem Biol. 2020. PMID: 32383857 Free PMC article.
-
Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties.Int J Mol Sci. 2021 Feb 5;22(4):1611. doi: 10.3390/ijms22041611. Int J Mol Sci. 2021. PMID: 33562633 Free PMC article. Review.
-
Macrocyclization of an all-d linear α-helical peptide imparts cellular permeability.Chem Sci. 2020 May 11;11(21):5577-5591. doi: 10.1039/c9sc06383h. eCollection 2020 Jun 7. Chem Sci. 2020. PMID: 32874502 Free PMC article.
-
Inhibition of calcium-triggered secretion by hydrocarbon-stapled peptides.Nature. 2022 Mar;603(7903):949-956. doi: 10.1038/s41586-022-04543-1. Epub 2022 Mar 23. Nature. 2022. PMID: 35322233 Free PMC article.
-
Will Peptides Help to Stop COVID-19?Biochemistry (Mosc). 2022 Jul;87(7):590-604. doi: 10.1134/S0006297922070021. Biochemistry (Mosc). 2022. PMID: 36154880 Free PMC article. Review.
References
-
- Craik D.J., Fairlie D.P., Liras S., Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. - PubMed
-
- Rosenblum D., Peer D. Omics-based nanomedicine: the future of personalized oncology. Cancer Lett. 2014;352:126–136. - PubMed
-
- Ruffner H., Bauer A., Bouwmeester T. Human protein–protein interaction networks and the value for drug discovery. Drug Discov Today. 2007;12:709–716. - PubMed
-
- Verdine G.L., Hilinski G.J. 1st ed. vol. 503. Elsevier Inc; 2012. Stapled peptides for intracellular drug targets. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials