

The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads
- PMID: 30783653
- PMCID: PMC6486549
- DOI: 10.1093/nar/gkz114
The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads
Abstract
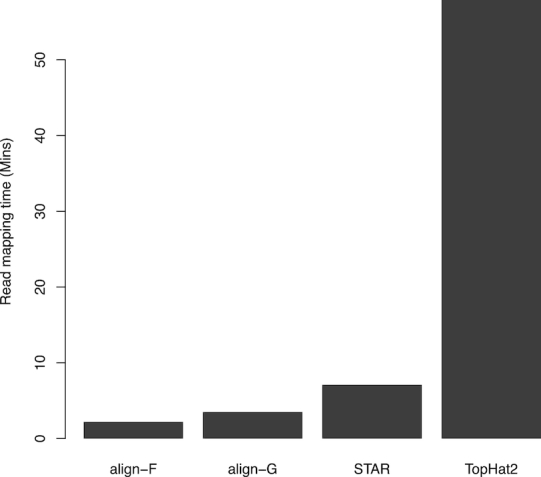
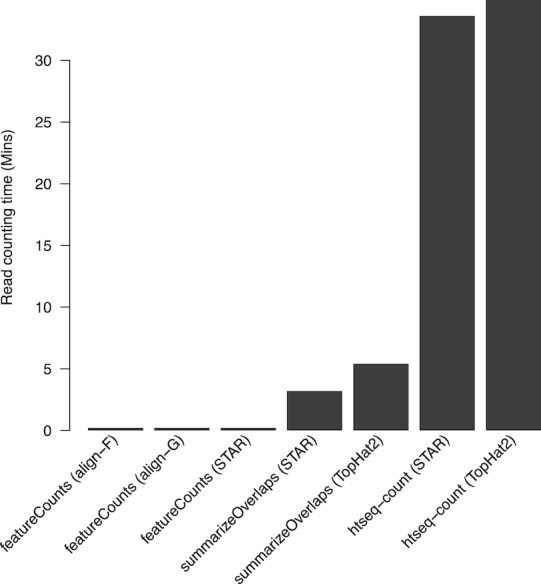
We present Rsubread, a Bioconductor software package that provides high-performance alignment and read counting functions for RNA-seq reads. Rsubread is based on the successful Subread suite with the added ease-of-use of the R programming environment, creating a matrix of read counts directly as an R object ready for downstream analysis. It integrates read mapping and quantification in a single package and has no software dependencies other than R itself. We demonstrate Rsubread's ability to detect exon-exon junctions de novo and to quantify expression at the level of either genes, exons or exon junctions. The resulting read counts can be input directly into a wide range of downstream statistical analyses using other Bioconductor packages. Using SEQC data and simulations, we compare Rsubread to TopHat2, STAR and HTSeq as well as to counting functions in the Bioconductor infrastructure packages. We consider the performance of these tools on the combined quantification task starting from raw sequence reads through to summary counts, and in particular evaluate the performance of different combinations of alignment and counting algorithms. We show that Rsubread is faster and uses less memory than competitor tools and produces read count summaries that more accurately correlate with true values.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.Bioinformatics. 2014 Apr 1;30(7):923-30. doi: 10.1093/bioinformatics/btt656. Epub 2013 Nov 13. Bioinformatics. 2014. PMID: 24227677
-
From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline.F1000Res. 2016 Jun 20;5:1438. doi: 10.12688/f1000research.8987.2. eCollection 2016. F1000Res. 2016. PMID: 27508061 Free PMC article.
-
It's DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR.Methods Mol Biol. 2016;1418:391-416. doi: 10.1007/978-1-4939-3578-9_19. Methods Mol Biol. 2016. PMID: 27008025
-
Mapping RNA-seq Reads with STAR.Curr Protoc Bioinformatics. 2015 Sep 3;51:11.14.1-11.14.19. doi: 10.1002/0471250953.bi1114s51. Curr Protoc Bioinformatics. 2015. PMID: 26334920 Free PMC article. Review.
-
Computational methods for transcriptome annotation and quantification using RNA-seq.Nat Methods. 2011 Jun;8(6):469-77. doi: 10.1038/nmeth.1613. Epub 2011 May 27. Nat Methods. 2011. PMID: 21623353 Review.
Cited by
-
Faster and more accurate assessment of differential transcript expression with Gibbs sampling and edgeR v4.NAR Genom Bioinform. 2024 Nov 4;6(4):lqae151. doi: 10.1093/nargab/lqae151. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39498433 Free PMC article.
-
Strain level and comprehensive microbiome analysis in inflammatory bowel disease via multi-technology meta-analysis identifies key bacterial influencers of disease.Front Microbiol. 2022 Oct 14;13:961020. doi: 10.3389/fmicb.2022.961020. eCollection 2022. Front Microbiol. 2022. PMID: 36312950 Free PMC article.
-
A human iPSC-array-based GWAS identifies a virus susceptibility locus in the NDUFA4 gene and functional variants.Cell Stem Cell. 2022 Oct 6;29(10):1475-1490.e6. doi: 10.1016/j.stem.2022.09.008. Cell Stem Cell. 2022. PMID: 36206731 Free PMC article.
-
An in vitro approach reveals molecular mechanisms underlying endocrine disruptor-induced epimutagenesis.Elife. 2024 Oct 3;13:RP93975. doi: 10.7554/eLife.93975. Elife. 2024. PMID: 39361026 Free PMC article.
-
Transcriptome analysis of primary adult B-cell lineage acute lymphoblastic leukemia identifies pathogenic variants and gene fusions, and predicts subtypes for in depth molecular diagnosis.Eur J Haematol. 2024 May;112(5):731-742. doi: 10.1111/ejh.14164. Epub 2024 Jan 8. Eur J Haematol. 2024. PMID: 38192186
References
-
- Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general-purpose read summarization program. Bioinformatics. 2014; 30:923–930. - PubMed
-
- Su Z., Labaj P.P., Li S., Thierry-Mieg J., Thierry-Mieg D., Shi W., Wang C., Schroth G.P., Setterquist R.A., Thompson J.F. et al. .. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014; 32:903–914. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
