

BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension
- PMID: 30759996
- PMCID: PMC6519484
- DOI: 10.1161/CIRCULATIONAHA.118.035889
BOLA (BolA Family Member 3) Deficiency Controls Endothelial Metabolism and Glycine Homeostasis in Pulmonary Hypertension
Abstract
Background: Deficiencies of iron-sulfur (Fe-S) clusters, metal complexes that control redox state and mitochondrial metabolism, have been linked to pulmonary hypertension (PH), a deadly vascular disease with poorly defined molecular origins. BOLA3 (BolA Family Member 3) regulates Fe-S biogenesis, and mutations in BOLA3 result in multiple mitochondrial dysfunction syndrome, a fatal disorder associated with PH. The mechanistic role of BOLA3 in PH remains undefined.
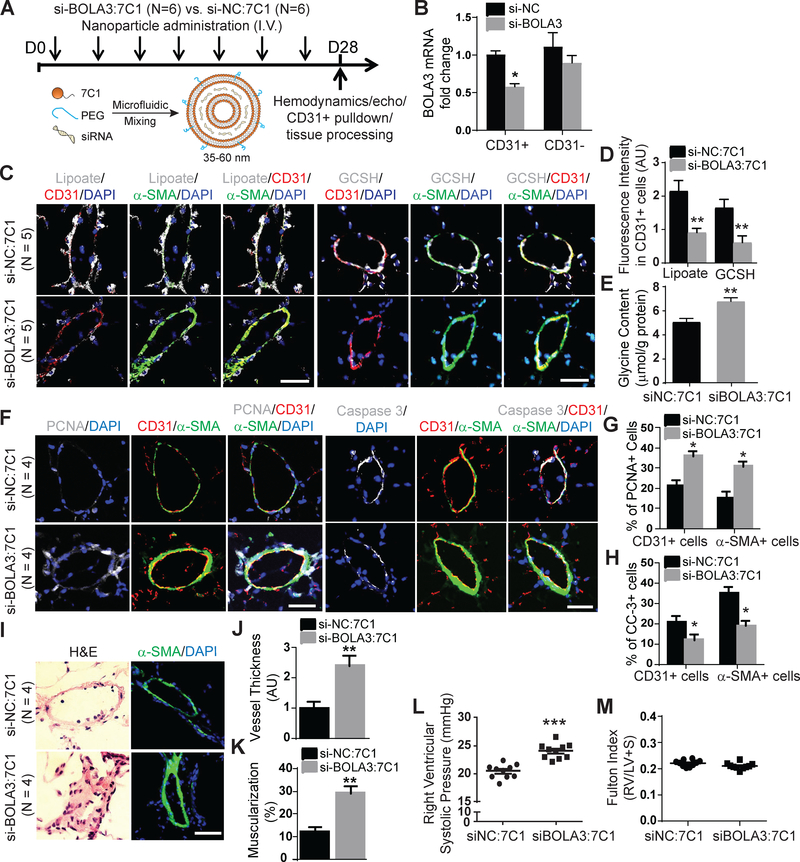
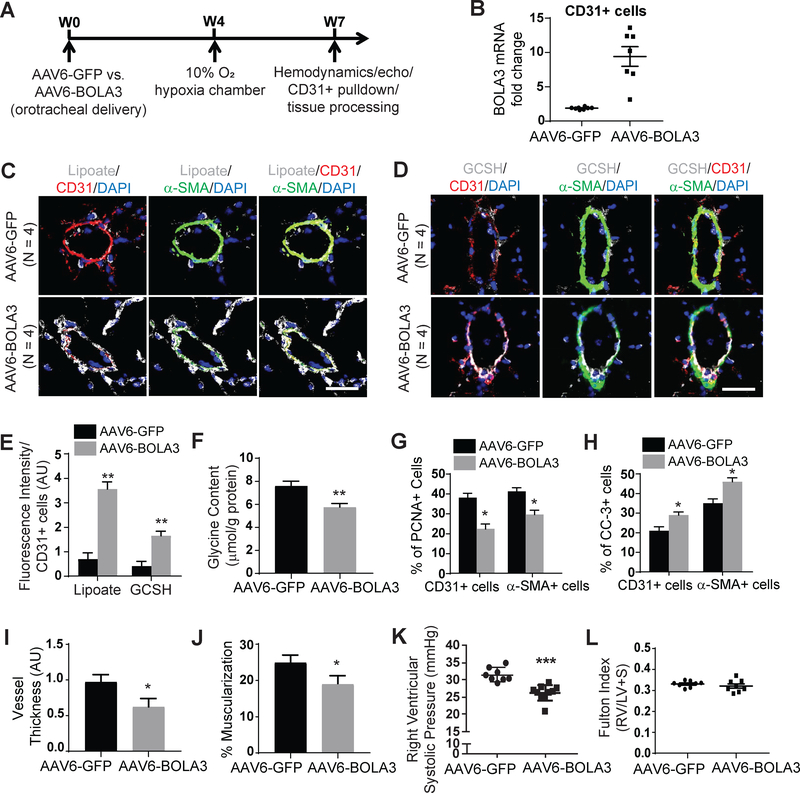
Methods: In vitro assessment of BOLA3 regulation and gain- and loss-of-function assays were performed in human pulmonary artery endothelial cells using siRNA and lentiviral vectors expressing the mitochondrial isoform of BOLA3. Polymeric nanoparticle 7C1 was used for lung endothelium-specific delivery of BOLA3 siRNA oligonucleotides in mice. Overexpression of pulmonary vascular BOLA3 was performed by orotracheal transgene delivery of adeno-associated virus in mouse models of PH.
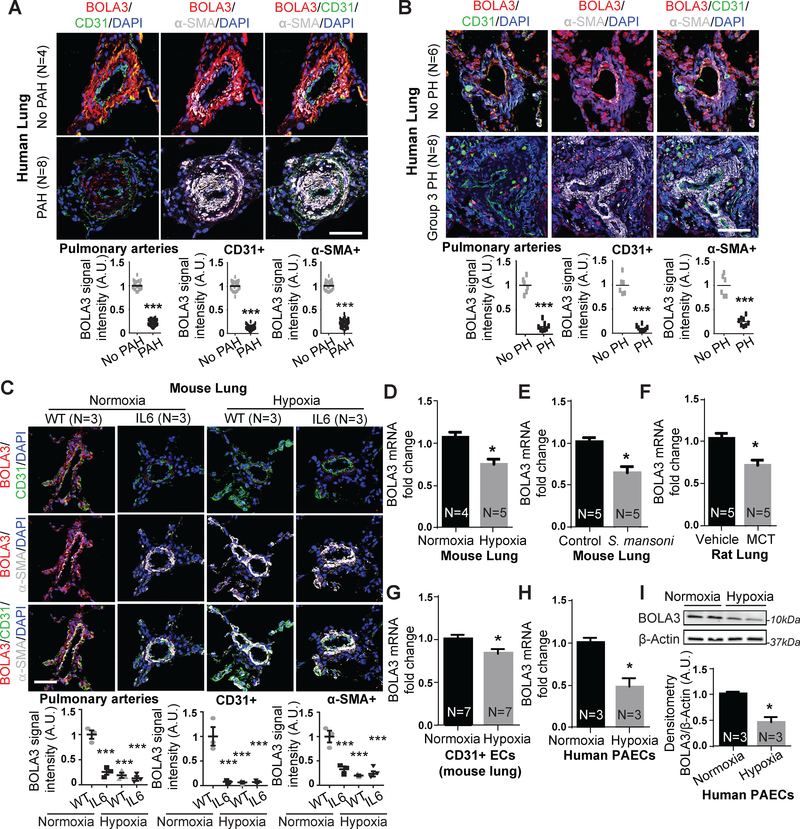
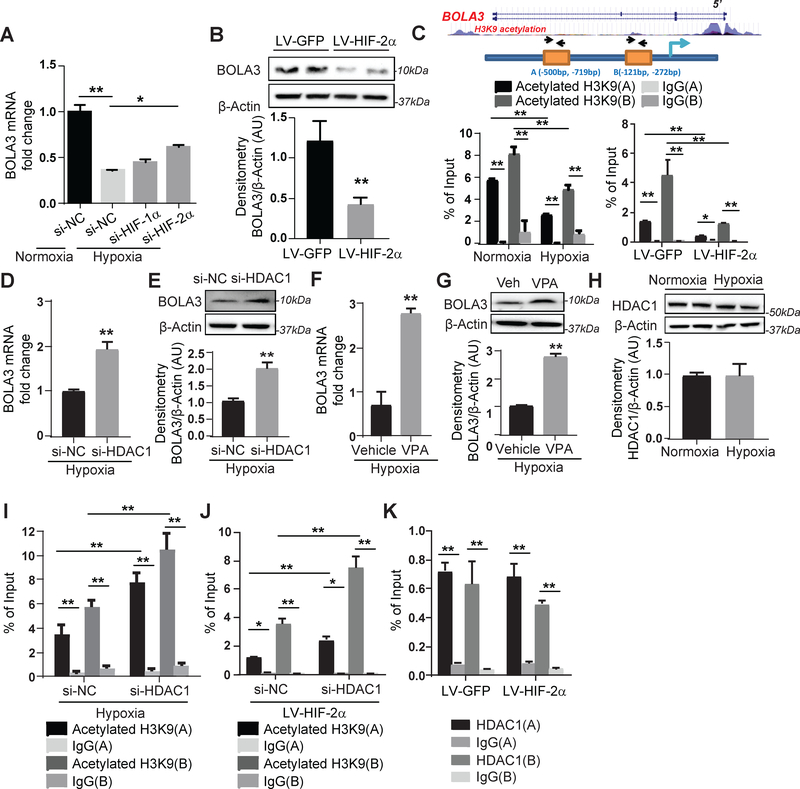
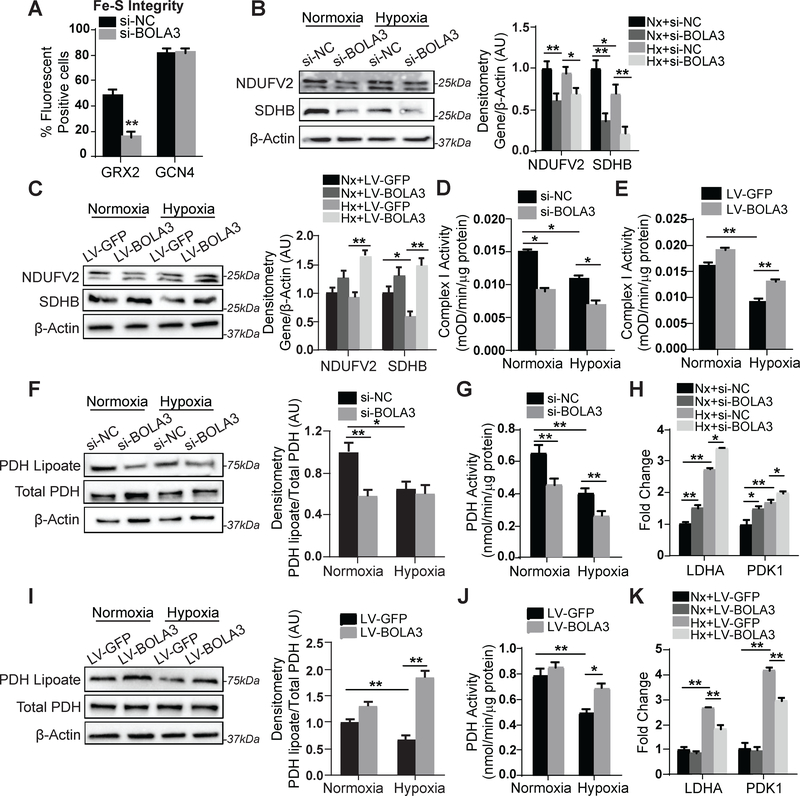
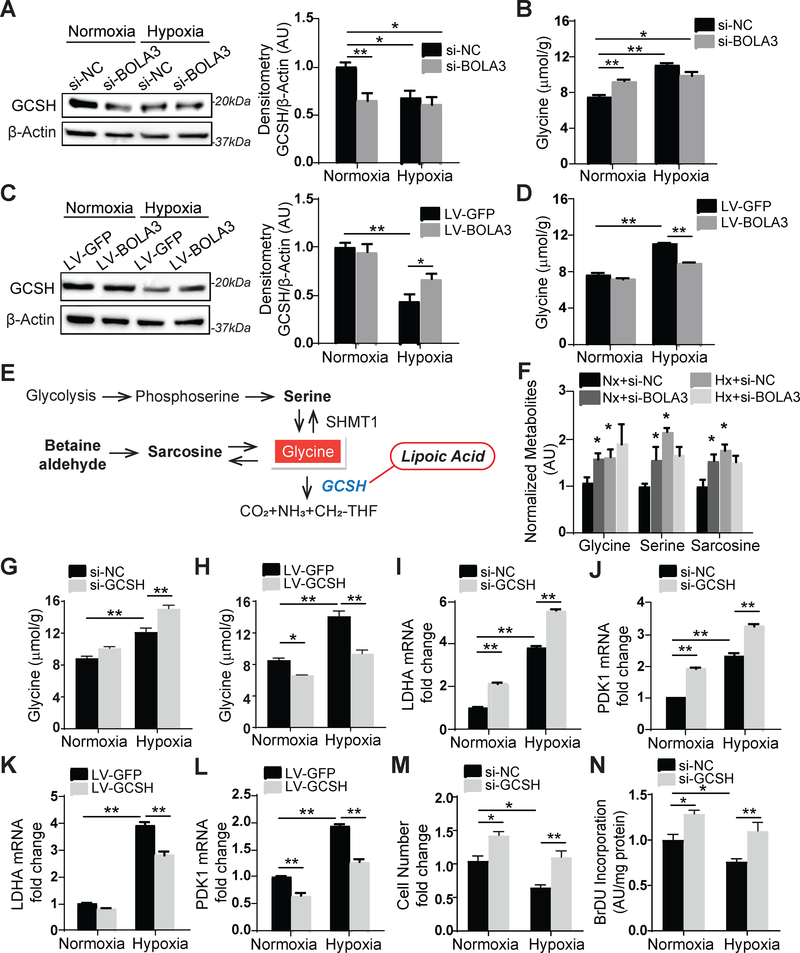
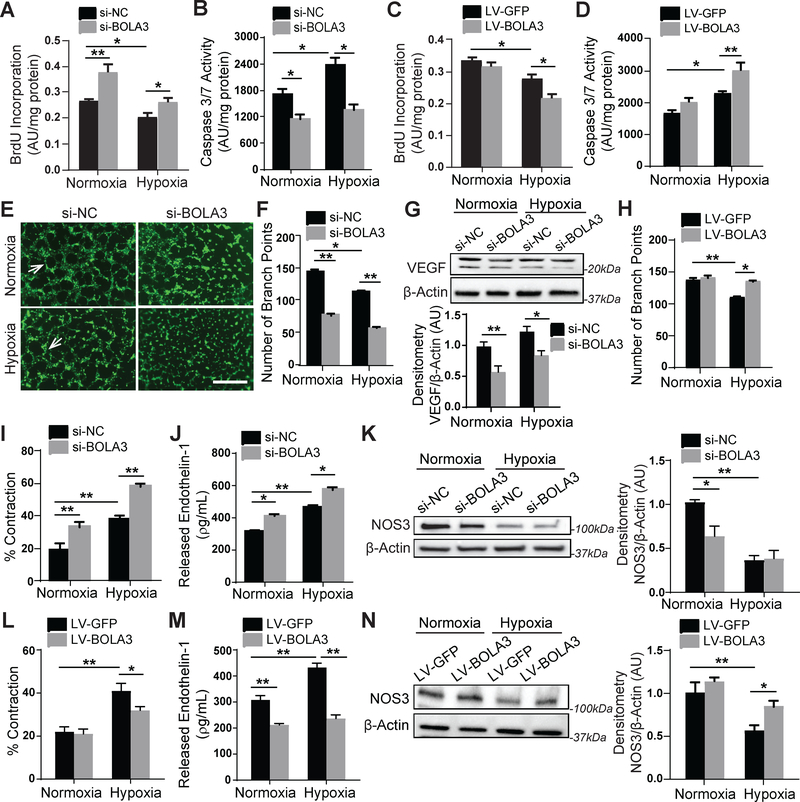
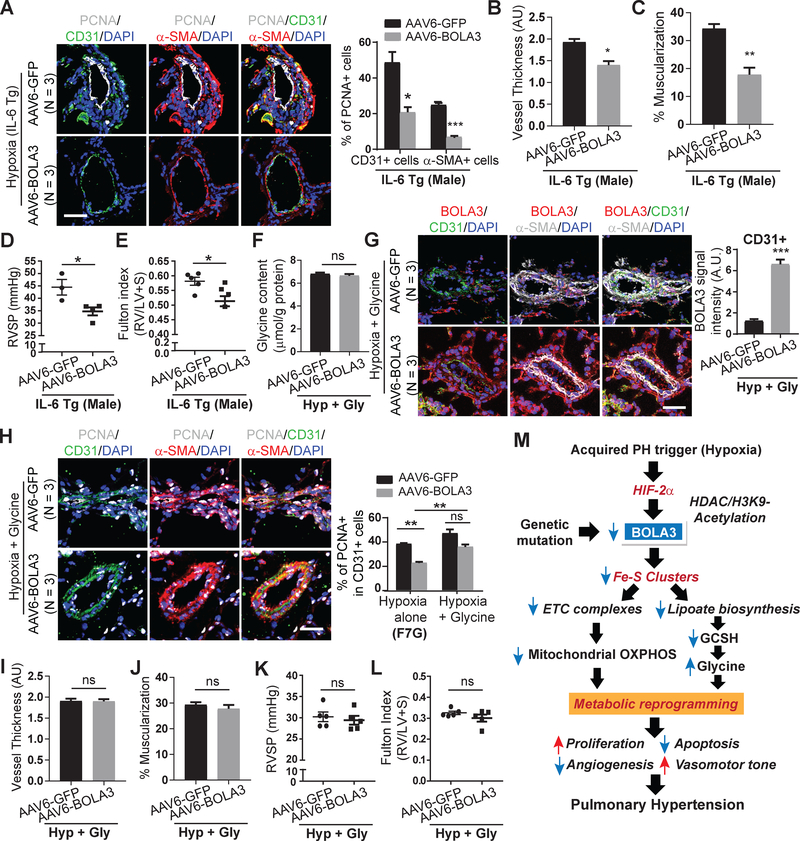
Results: In cultured hypoxic pulmonary artery endothelial cells, lung from human patients with Group 1 and 3 PH, and multiple rodent models of PH, endothelial BOLA3 expression was downregulated, which involved hypoxia inducible factor-2α-dependent transcriptional repression via histone deacetylase 1-mediated histone deacetylation. In vitro gain- and loss-of-function studies demonstrated that BOLA3 regulated Fe-S integrity, thus modulating lipoate-containing 2-oxoacid dehydrogenases with consequent control over glycolysis and mitochondrial respiration. In contexts of siRNA knockdown and naturally occurring human genetic mutation, cellular BOLA3 deficiency downregulated the glycine cleavage system protein H, thus bolstering intracellular glycine content. In the setting of these alterations of oxidative metabolism and glycine levels, BOLA3 deficiency increased endothelial proliferation, survival, and vasoconstriction while decreasing angiogenic potential. In vivo, pharmacological knockdown of endothelial BOLA3 and targeted overexpression of BOLA3 in mice demonstrated that BOLA3 deficiency promotes histological and hemodynamic manifestations of PH. Notably, the therapeutic effects of BOLA3 expression were reversed by exogenous glycine supplementation.
Conclusions: BOLA3 acts as a crucial lynchpin connecting Fe-S-dependent oxidative respiration and glycine homeostasis with endothelial metabolic reprogramming critical to PH pathogenesis. These results provide a molecular explanation for the clinical associations linking PH with hyperglycinemic syndromes and mitochondrial disorders. These findings also identify novel metabolic targets, including those involved in epigenetics, Fe-S biogenesis, and glycine biology, for diagnostic and therapeutic development.
Keywords: endothelium; glycine; hypertension, pulmonary; mitochondria.
Conflict of interest statement
Disclosures
S.Y.C. has served as a consultant for Zogenix (Significant) and Vivus (Modest) and holds research grants for Pfizer and Actelion. The authors declare no other conflicts of interest.
Figures
Comment in
-
Does Glycine cataBOLAsm Drive Pulmonary Hypertension?Circulation. 2019 May 7;139(19):2256-2259. doi: 10.1161/CIRCULATIONAHA.119.039611. Circulation. 2019. PMID: 31059318 No abstract available.
Similar articles
-
Bola3 Regulates Beige Adipocyte Thermogenesis via Maintaining Mitochondrial Homeostasis and Lipolysis.Front Endocrinol (Lausanne). 2021 Jan 11;11:592154. doi: 10.3389/fendo.2020.592154. eCollection 2020. Front Endocrinol (Lausanne). 2021. PMID: 33505355 Free PMC article.
-
Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension.EMBO Mol Med. 2015 Jun;7(6):695-713. doi: 10.15252/emmm.201404511. EMBO Mol Med. 2015. PMID: 25825391 Free PMC article.
-
Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes.Am J Hum Genet. 2011 Oct 7;89(4):486-95. doi: 10.1016/j.ajhg.2011.08.011. Epub 2011 Sep 22. Am J Hum Genet. 2011. PMID: 21944046 Free PMC article.
-
Mitochondrial iron-sulfur protein biogenesis and human disease.Biochimie. 2014 May;100:61-77. doi: 10.1016/j.biochi.2014.01.010. Epub 2014 Jan 23. Biochimie. 2014. PMID: 24462711 Review.
-
Mitochondrial and Metabolic Drivers of Pulmonary Vascular Endothelial Dysfunction in Pulmonary Hypertension.Adv Exp Med Biol. 2017;967:373-383. doi: 10.1007/978-3-319-63245-2_24. Adv Exp Med Biol. 2017. PMID: 29047100 Free PMC article. Review.
Cited by
-
Double-edge sword roles of iron in driving energy production versus instigating ferroptosis.Cell Death Dis. 2022 Jan 10;13(1):40. doi: 10.1038/s41419-021-04490-1. Cell Death Dis. 2022. PMID: 35013137 Free PMC article. Review.
-
Endothelial Cell Mechano-Metabolomic Coupling to Disease States in the Lung Microvasculature.Front Bioeng Biotechnol. 2019 Jul 19;7:172. doi: 10.3389/fbioe.2019.00172. eCollection 2019. Front Bioeng Biotechnol. 2019. PMID: 31380363 Free PMC article. Review.
-
Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins.Curr Opin Chem Biol. 2020 Apr;55:34-44. doi: 10.1016/j.cbpa.2019.11.014. Epub 2020 Jan 6. Curr Opin Chem Biol. 2020. PMID: 31918395 Free PMC article. Review.
-
Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis.Trends Biochem Sci. 2020 May;45(5):411-426. doi: 10.1016/j.tibs.2020.02.001. Epub 2020 Mar 6. Trends Biochem Sci. 2020. PMID: 32311335 Free PMC article. Review.
-
Schistosomiasis Pulmonary Arterial Hypertension.Front Immunol. 2020 Dec 10;11:608883. doi: 10.3389/fimmu.2020.608883. eCollection 2020. Front Immunol. 2020. PMID: 33362796 Free PMC article. Review.
References
-
- White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, Perrella MA, Osorio JC, Haley KJ, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Khan OF, Bader A, Gochuico BR, Matar M, Polach K, Johannessen NM, Prosser HM, Anderson DG, Langer R, Zweier JL, Bindoff LA, Systrom D, Waxman AB, Jin RC and Chan SY. Genetic and hypoxic alterations of the microRNA-210-ISCU½ axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med. 2015; 7:695–713. - PMC - PubMed
-
- Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, Ogilvie I, Shoubridge EA and Robinson BH. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011; 89:486–495. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
