

PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress
- PMID: 30700906
- PMCID: PMC6426636
- DOI: 10.1038/s41586-019-0895-y
PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress
Abstract
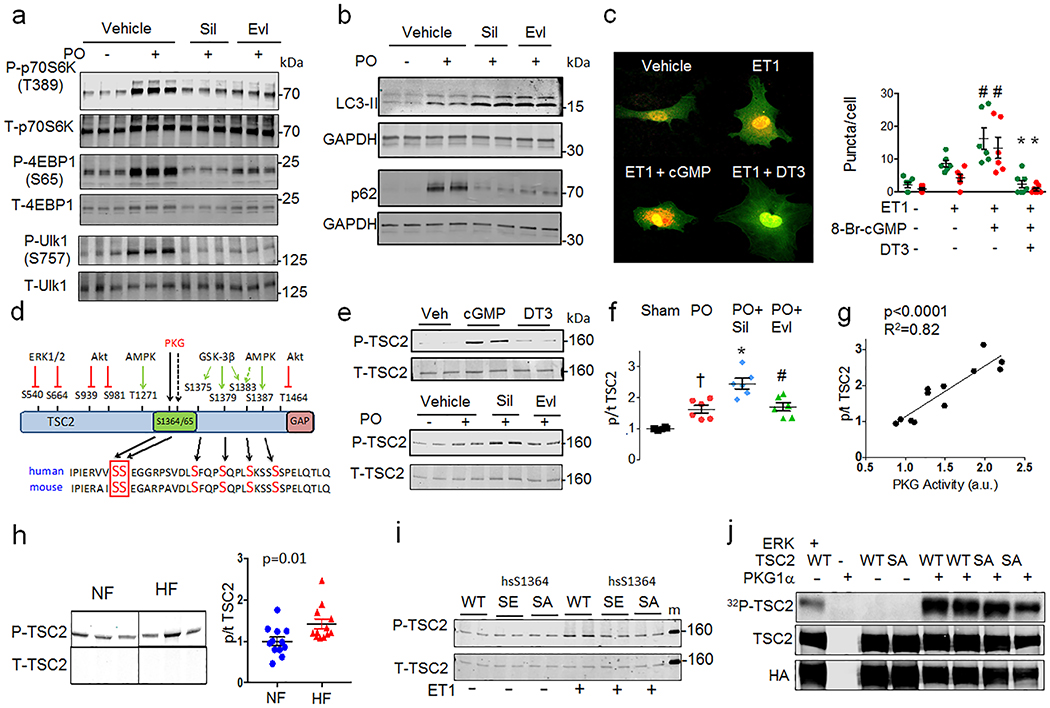
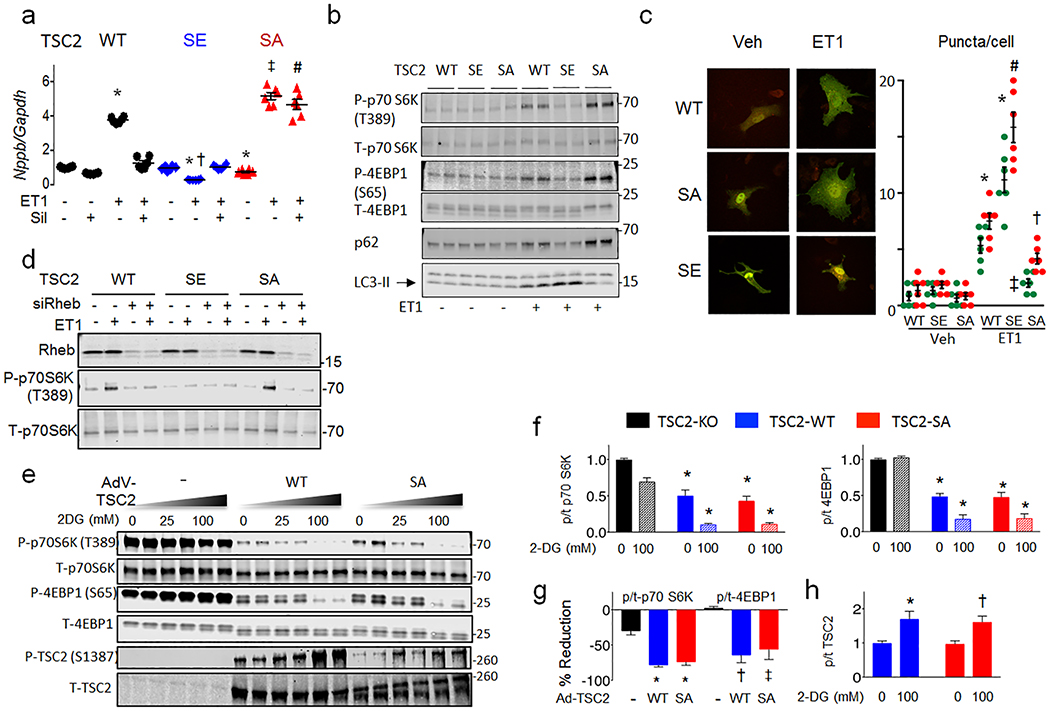
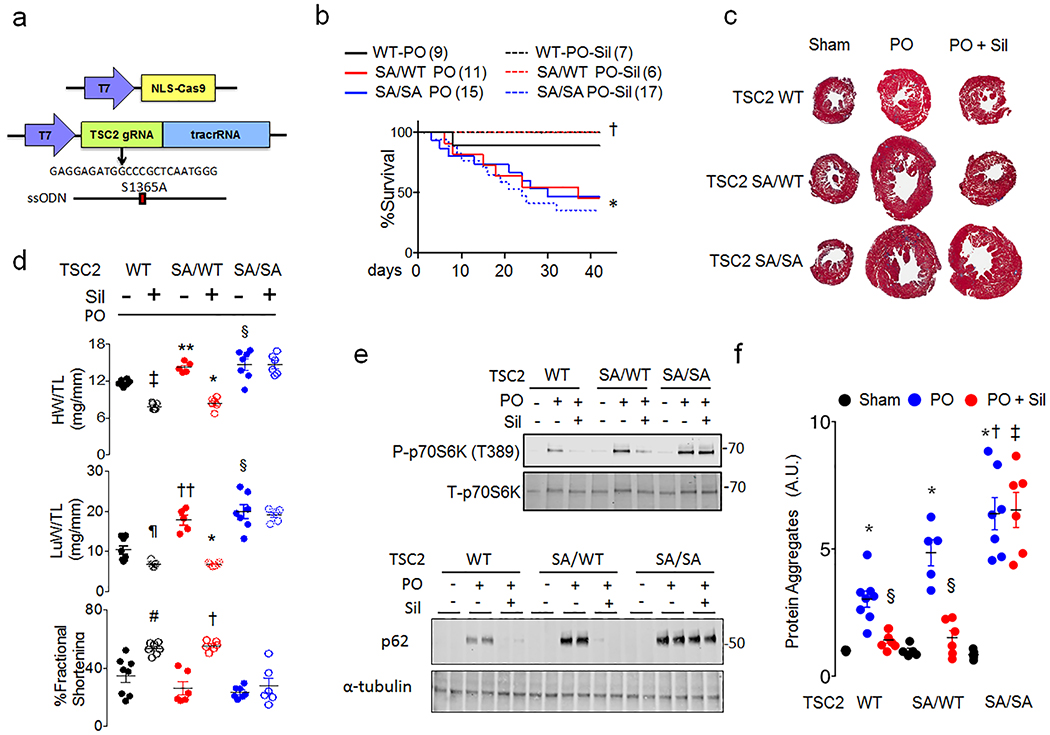
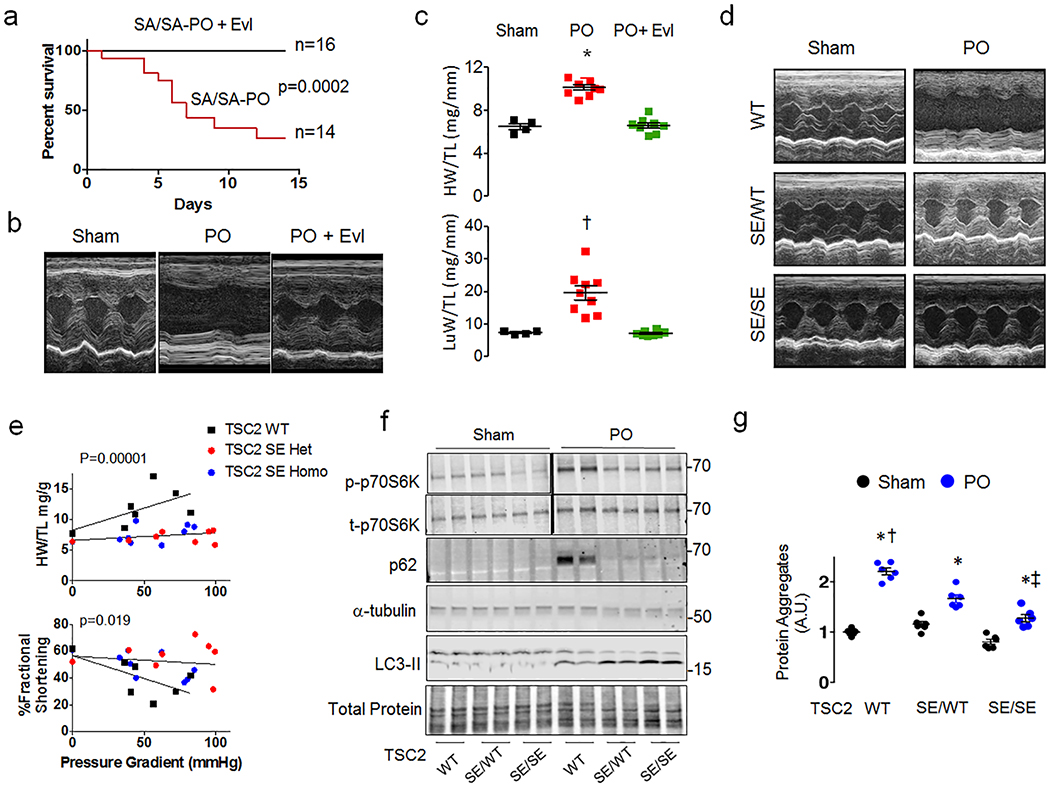
The mechanistic target of rapamycin complex-1 (mTORC1) coordinates regulation of growth, metabolism, protein synthesis and autophagy1. Its hyperactivation contributes to disease in numerous organs, including the heart1,2, although broad inhibition of mTORC1 risks interference with its homeostatic roles. Tuberin (TSC2) is a GTPase-activating protein and prominent intrinsic regulator of mTORC1 that acts through modulation of RHEB (Ras homologue enriched in brain). TSC2 constitutively inhibits mTORC1; however, this activity is modified by phosphorylation from multiple signalling kinases that in turn inhibits (AMPK and GSK-3β) or stimulates (AKT, ERK and RSK-1) mTORC1 activity3-9. Each kinase requires engagement of multiple serines, impeding analysis of their role in vivo. Here we show that phosphorylation or gain- or loss-of-function mutations at either of two adjacent serine residues in TSC2 (S1365 and S1366 in mice; S1364 and S1365 in humans) can bidirectionally control mTORC1 activity stimulated by growth factors or haemodynamic stress, and consequently modulate cell growth and autophagy. However, basal mTORC1 activity remains unchanged. In the heart, or in isolated cardiomyocytes or fibroblasts, protein kinase G1 (PKG1) phosphorylates these TSC2 sites. PKG1 is a primary effector of nitric oxide and natriuretic peptide signalling, and protects against heart disease10-13. Suppression of hypertrophy and stimulation of autophagy in cardiomyocytes by PKG1 requires TSC2 phosphorylation. Homozygous knock-in mice that express a phosphorylation-silencing mutation in TSC2 (TSC2(S1365A)) develop worse heart disease and have higher mortality after sustained pressure overload of the heart, owing to mTORC1 hyperactivity that cannot be rescued by PKG1 stimulation. However, cardiac disease is reduced and survival of heterozygote Tsc2S1365A knock-in mice subjected to the same stress is improved by PKG1 activation or expression of a phosphorylation-mimicking mutation (TSC2(S1365E)). Resting mTORC1 activity is not altered in either knock-in model. Therefore, TSC2 phosphorylation is both required and sufficient for PKG1-mediated cardiac protection against pressure overload. The serine residues identified here provide a genetic tool for bidirectional regulation of the amplitude of stress-stimulated mTORC1 activity.
Figures
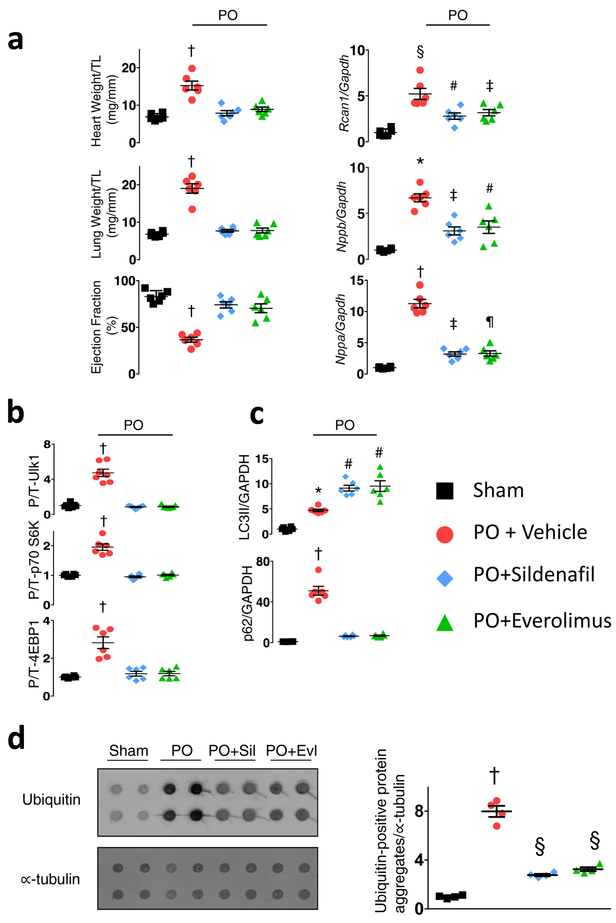
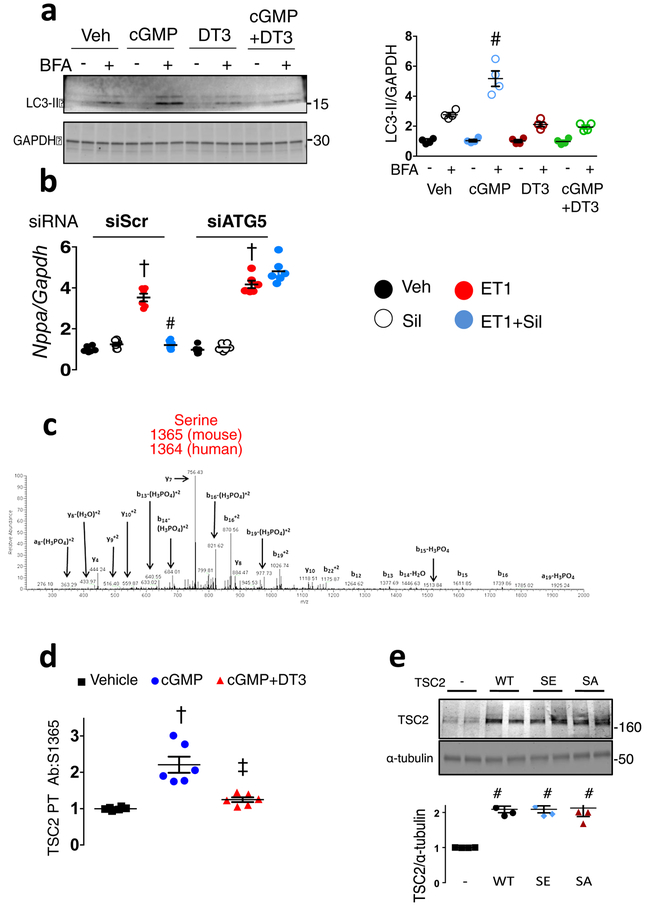
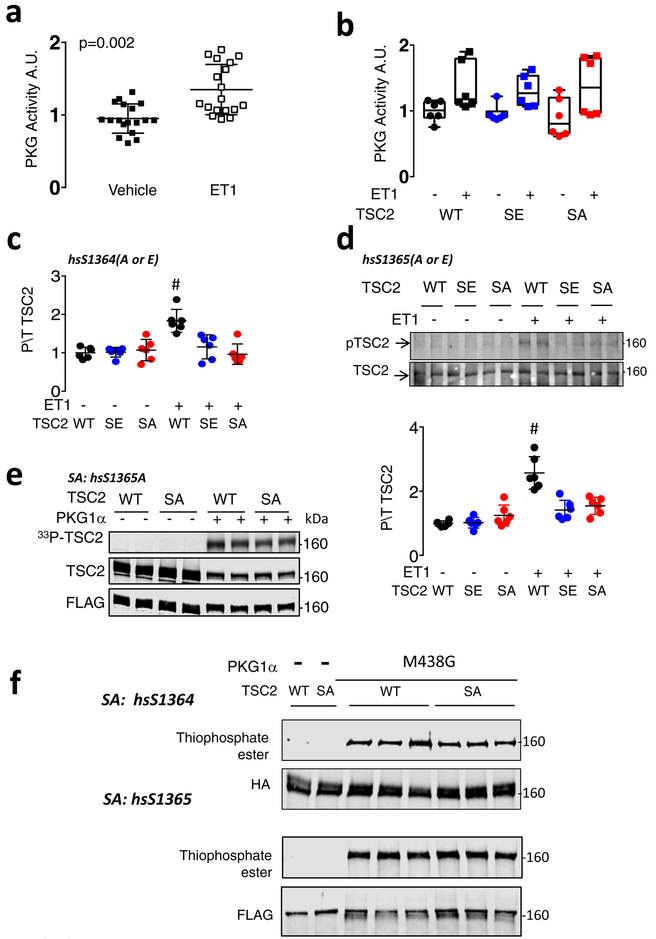
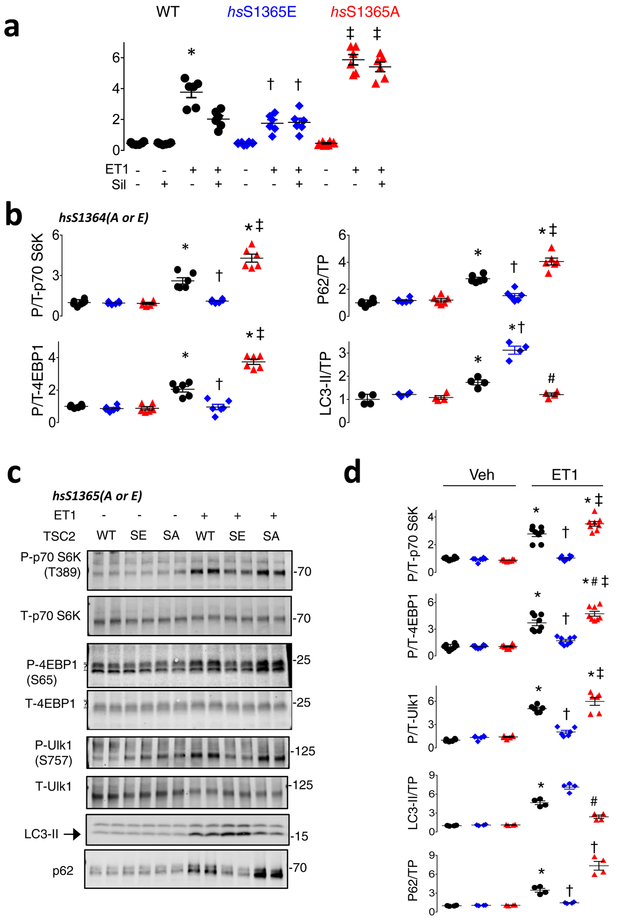
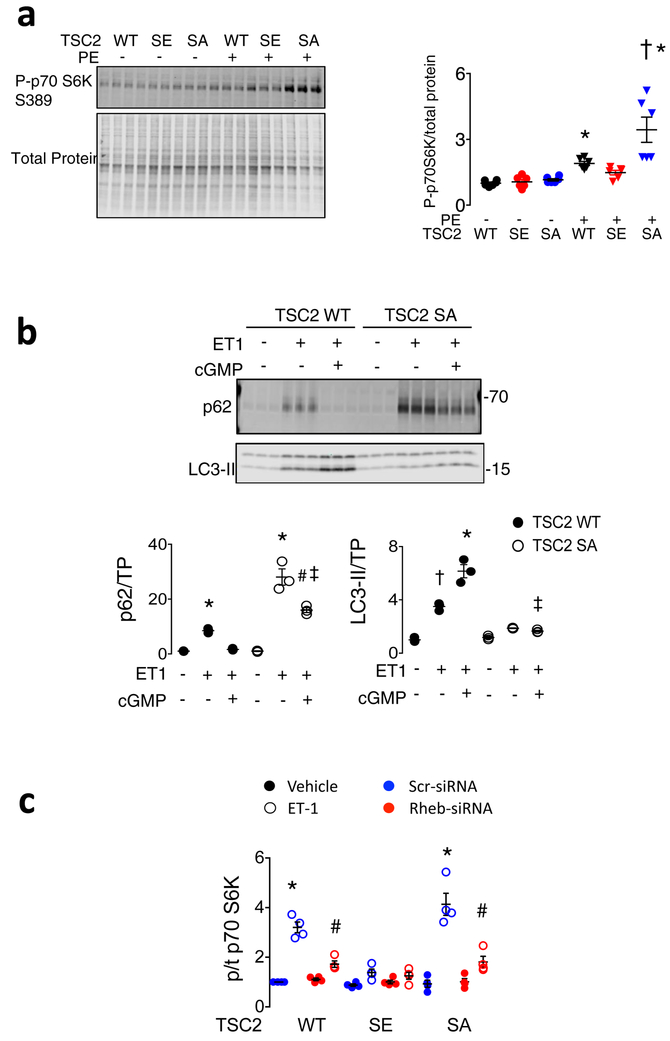
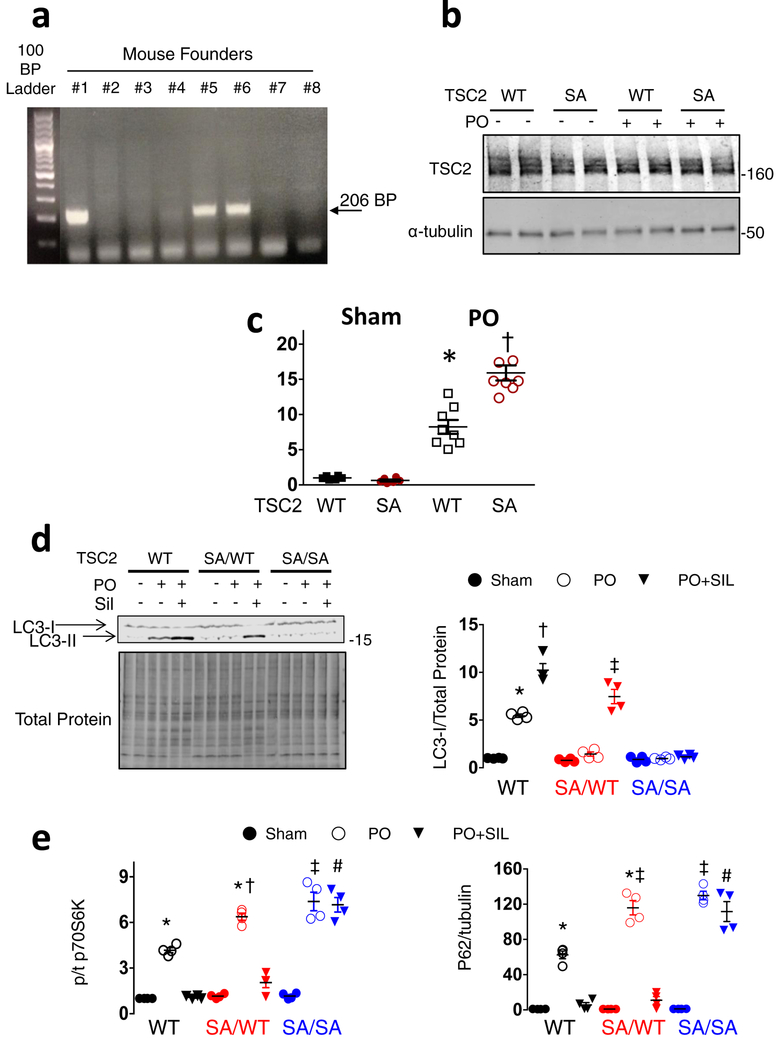
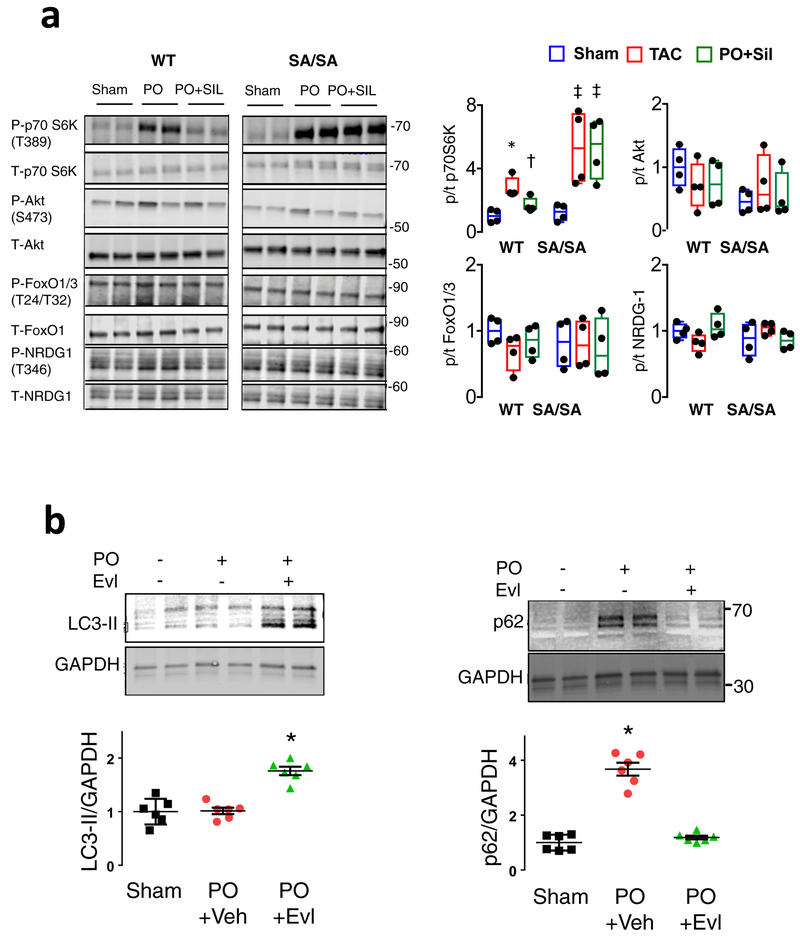
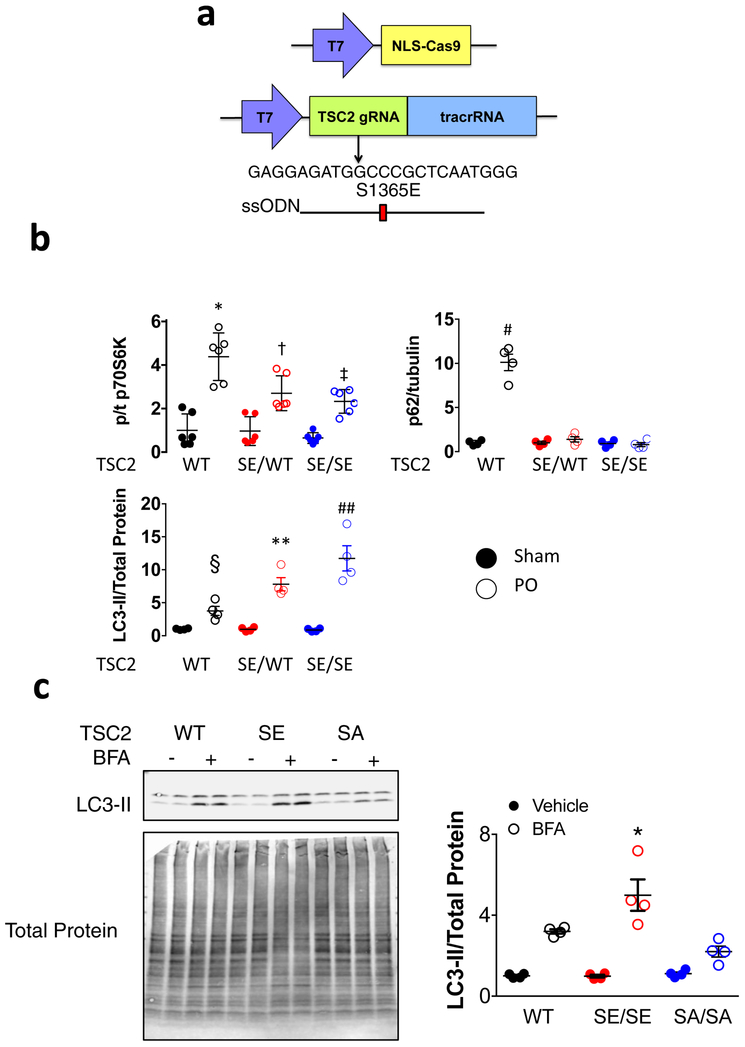
Comment in
-
Signalling protein protects the heart muscle from pressure-related stress.Nature. 2019 Feb;566(7743):187-188. doi: 10.1038/d41586-019-00245-3. Nature. 2019. PMID: 30737501 No abstract available.
Similar articles
-
PKG1α Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy.Circ Res. 2020 Jul 31;127(4):522-533. doi: 10.1161/CIRCRESAHA.119.315714. Epub 2020 May 12. Circ Res. 2020. PMID: 32393148 Free PMC article.
-
MTORC1-Regulated Metabolism Controlled by TSC2 Limits Cardiac Reperfusion Injury.Circ Res. 2021 Mar 5;128(5):639-651. doi: 10.1161/CIRCRESAHA.120.317710. Epub 2021 Jan 6. Circ Res. 2021. PMID: 33401933 Free PMC article.
-
Single serine on TSC2 exerts biased control over mTORC1 activation mediated by ERK1/2 but not Akt.Life Sci Alliance. 2022 Mar 14;5(6):e202101169. doi: 10.26508/lsa.202101169. Print 2022 Jun. Life Sci Alliance. 2022. PMID: 35288456 Free PMC article.
-
A complex interplay between Akt, TSC2 and the two mTOR complexes.Biochem Soc Trans. 2009 Feb;37(Pt 1):217-22. doi: 10.1042/BST0370217. Biochem Soc Trans. 2009. PMID: 19143635 Free PMC article. Review.
-
Genetic and Environmental Contributions to Autism Spectrum Disorder Through Mechanistic Target of Rapamycin.Biol Psychiatry Glob Open Sci. 2021 Sep 1;2(2):95-105. doi: 10.1016/j.bpsgos.2021.08.005. eCollection 2022 Apr. Biol Psychiatry Glob Open Sci. 2021. PMID: 36325164 Free PMC article. Review.
Cited by
-
Dietary carbohydrates restriction inhibits the development of cardiac hypertrophy and heart failure.Cardiovasc Res. 2021 Sep 28;117(11):2365-2376. doi: 10.1093/cvr/cvaa298. Cardiovasc Res. 2021. PMID: 33070172 Free PMC article.
-
The role of autophagy in cardiovascular pathology.Cardiovasc Res. 2022 Mar 16;118(4):934-950. doi: 10.1093/cvr/cvab158. Cardiovasc Res. 2022. PMID: 33956077 Free PMC article. Review.
-
Autophagy in striated muscle diseases.Front Cardiovasc Med. 2022 Oct 13;9:1000067. doi: 10.3389/fcvm.2022.1000067. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36312227 Free PMC article. Review.
-
Reversible Treatment of Pressure Overload-Induced Left Ventricular Hypertrophy through Drd5 Nucleic Acid Delivery Mediated by Functional Polyaminoglycoside.Adv Sci (Weinh). 2021 Jan 6;8(5):2003706. doi: 10.1002/advs.202003706. eCollection 2021 Mar. Adv Sci (Weinh). 2021. PMID: 33717857 Free PMC article.
-
Targeting Protein Kinase G to Treat Cardiac Proteotoxicity.Front Physiol. 2020 Jul 28;11:858. doi: 10.3389/fphys.2020.00858. eCollection 2020. Front Physiol. 2020. PMID: 32848832 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
