

Chromatin features constrain structural variation across evolutionary timescales
- PMID: 30659153
- PMCID: PMC6369792
- DOI: 10.1073/pnas.1808631116
Chromatin features constrain structural variation across evolutionary timescales
Abstract
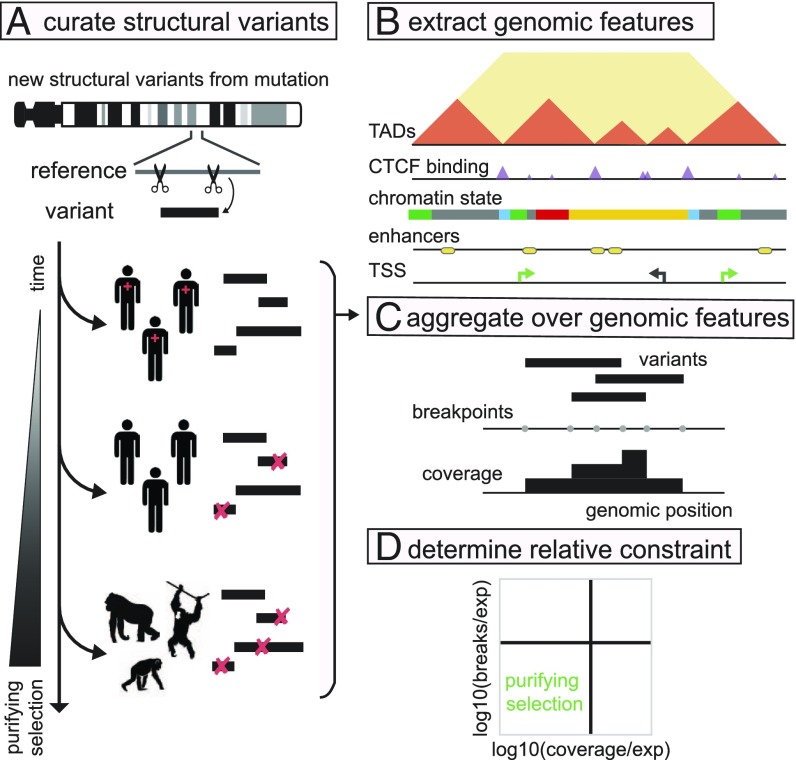
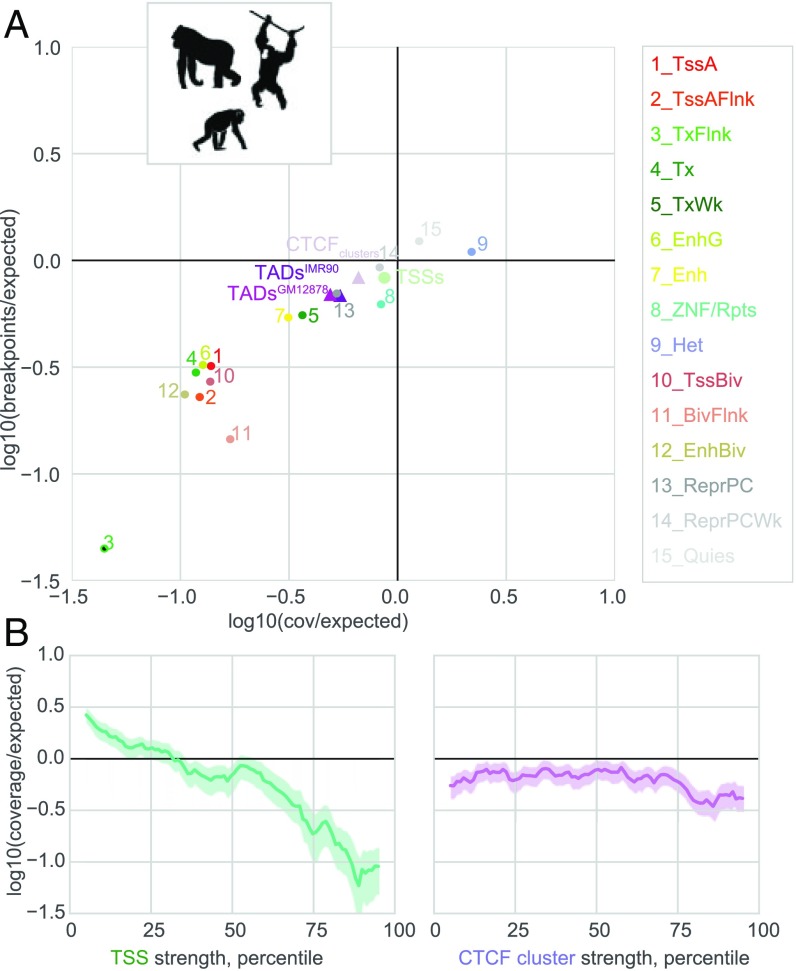
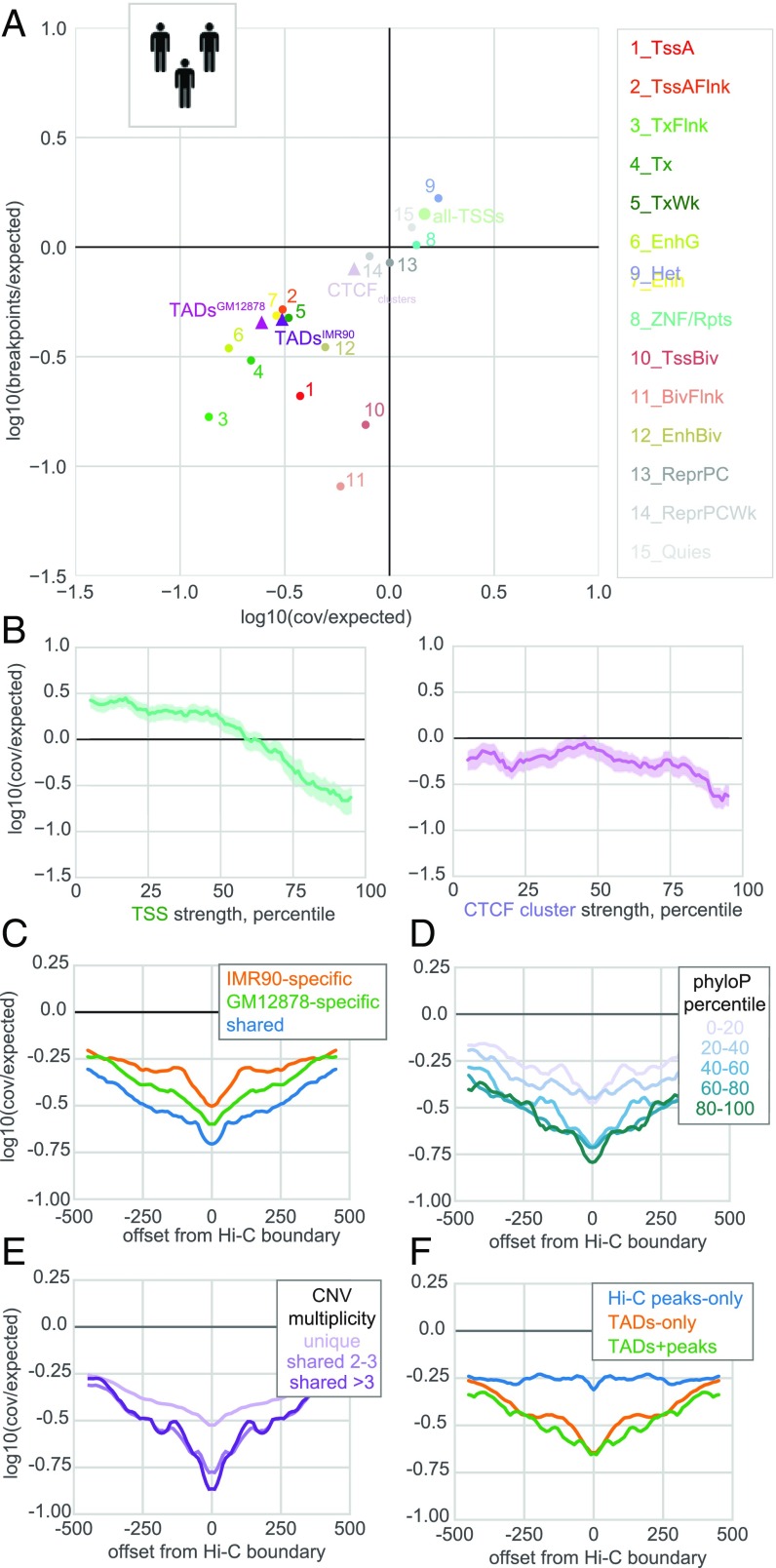
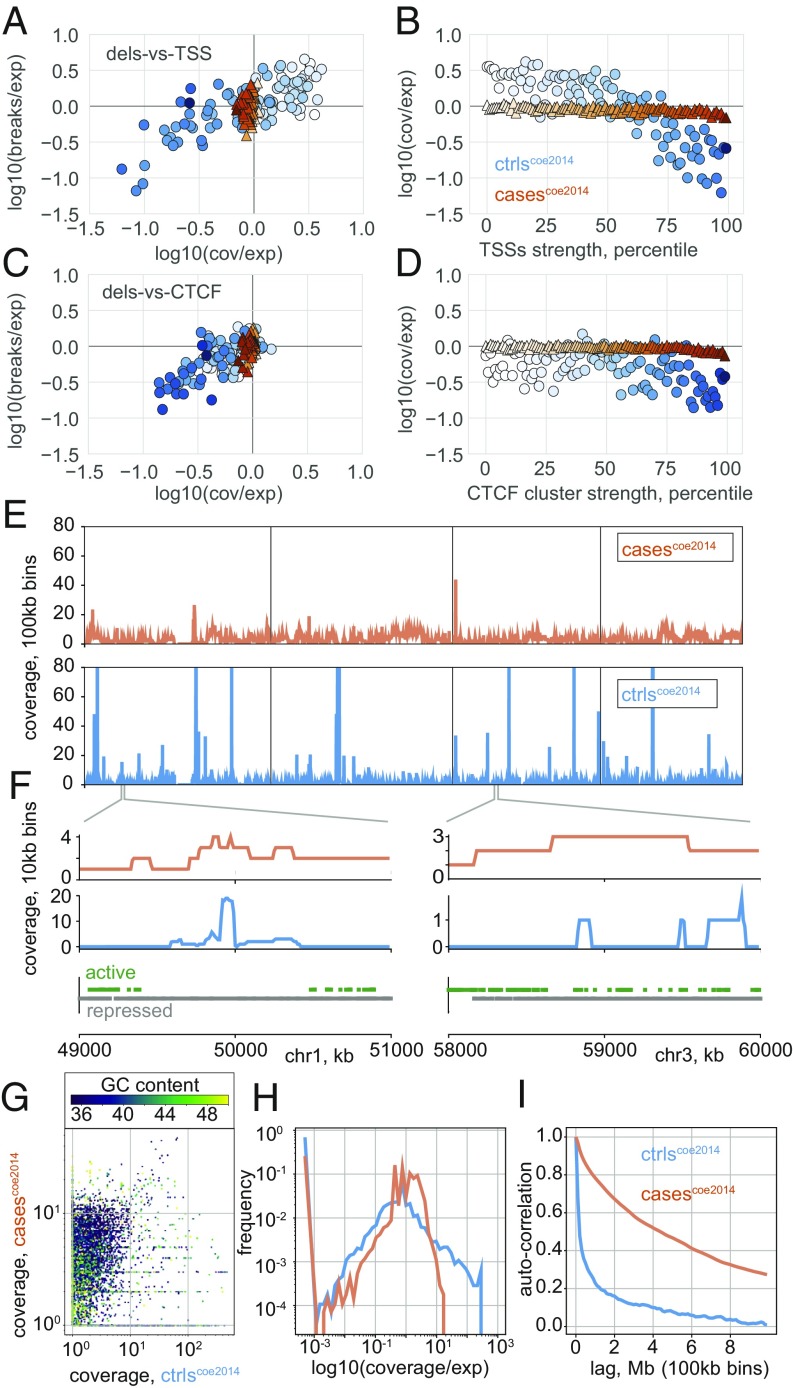
The potential impact of structural variants includes not only the duplication or deletion of coding sequences, but also the perturbation of noncoding DNA regulatory elements and structural chromatin features, including topological domains (TADs). Structural variants disrupting TAD boundaries have been implicated both in cancer and developmental disease; this likely occurs via "enhancer hijacking," whereby removal of the TAD boundary exposes enhancers to new target transcription start sites (TSSs). With this functional role, we hypothesized that boundaries would display evidence for negative selection. Here we demonstrate that the chromatin landscape constrains structural variation both within healthy humans and across primate evolution. In contrast, in patients with developmental delay, variants occur remarkably uniformly across genomic features, suggesting a potentially broad role for enhancer hijacking in human disease.
Keywords: CTCF; Hi-C; chromatin; comparative genomics; evolution.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Contribution of transposable elements and distal enhancers to evolution of human-specific features of interphase chromatin architecture in embryonic stem cells.Chromosome Res. 2018 Mar;26(1-2):61-84. doi: 10.1007/s10577-018-9571-6. Epub 2018 Jan 15. Chromosome Res. 2018. PMID: 29335803
-
Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders.Proc Natl Acad Sci U S A. 2015 Jun 16;112(24):7542-7. doi: 10.1073/pnas.1505463112. Epub 2015 Jun 1. Proc Natl Acad Sci U S A. 2015. PMID: 26034287 Free PMC article.
-
Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability.Am J Hum Genet. 2021 Feb 4;108(2):269-283. doi: 10.1016/j.ajhg.2021.01.001. Am J Hum Genet. 2021. PMID: 33545030 Free PMC article.
-
When TADs go bad: chromatin structure and nuclear organisation in human disease.F1000Res. 2017 Mar 24;6:F1000 Faculty Rev-314. doi: 10.12688/f1000research.10792.1. eCollection 2017. F1000Res. 2017. PMID: 28408976 Free PMC article. Review.
-
The connection between BRG1, CTCF and topoisomerases at TAD boundaries.Nucleus. 2017 Mar 4;8(2):150-155. doi: 10.1080/19491034.2016.1276145. Epub 2017 Jan 6. Nucleus. 2017. PMID: 28060558 Free PMC article. Review.
Cited by
-
Abnormal Chromatin Folding in the Molecular Pathogenesis of Epilepsy and Autism Spectrum Disorder: a Meta-synthesis with Systematic Searching.Mol Neurobiol. 2023 Feb;60(2):768-779. doi: 10.1007/s12035-022-03106-9. Epub 2022 Nov 11. Mol Neurobiol. 2023. PMID: 36367658 Free PMC article. Review.
-
Determining cellular CTCF and cohesin abundances to constrain 3D genome models.Elife. 2019 Jun 17;8:e40164. doi: 10.7554/eLife.40164. Elife. 2019. PMID: 31205001 Free PMC article.
-
Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing.Genes (Basel). 2020 Mar 4;11(3):276. doi: 10.3390/genes11030276. Genes (Basel). 2020. PMID: 32143403 Free PMC article.
-
Machine learning reveals the diversity of human 3D chromatin contact patterns.bioRxiv [Preprint]. 2023 Dec 23:2023.12.22.573104. doi: 10.1101/2023.12.22.573104. bioRxiv. 2023. Update in: Mol Biol Evol. 2024 Oct 4;41(10):msae209. doi: 10.1093/molbev/msae209 PMID: 38187606 Free PMC article. Updated. Preprint.
-
Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain?Genes (Basel). 2019 Jun 7;10(6):432. doi: 10.3390/genes10060432. Genes (Basel). 2019. PMID: 31181654 Free PMC article. Review.
References
-
- Cheng Z, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. - PubMed
-
- Krijger PHL, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol. 2016;17:771–782. - PubMed
-
- Spielmann M, Mundlos S. Looking beyond the genes: The role of non-coding variants in human disease. Hum Mol Genet. 2016;25:R157–R165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
