

Genetically Encoding Albumin Binding into Chemotherapeutic-loaded Polypeptide Nanoparticles Enhances Their Antitumor Efficacy
- PMID: 30461287
- PMCID: PMC10387222
- DOI: 10.1021/acs.nanolett.8b03558
Genetically Encoding Albumin Binding into Chemotherapeutic-loaded Polypeptide Nanoparticles Enhances Their Antitumor Efficacy
Abstract
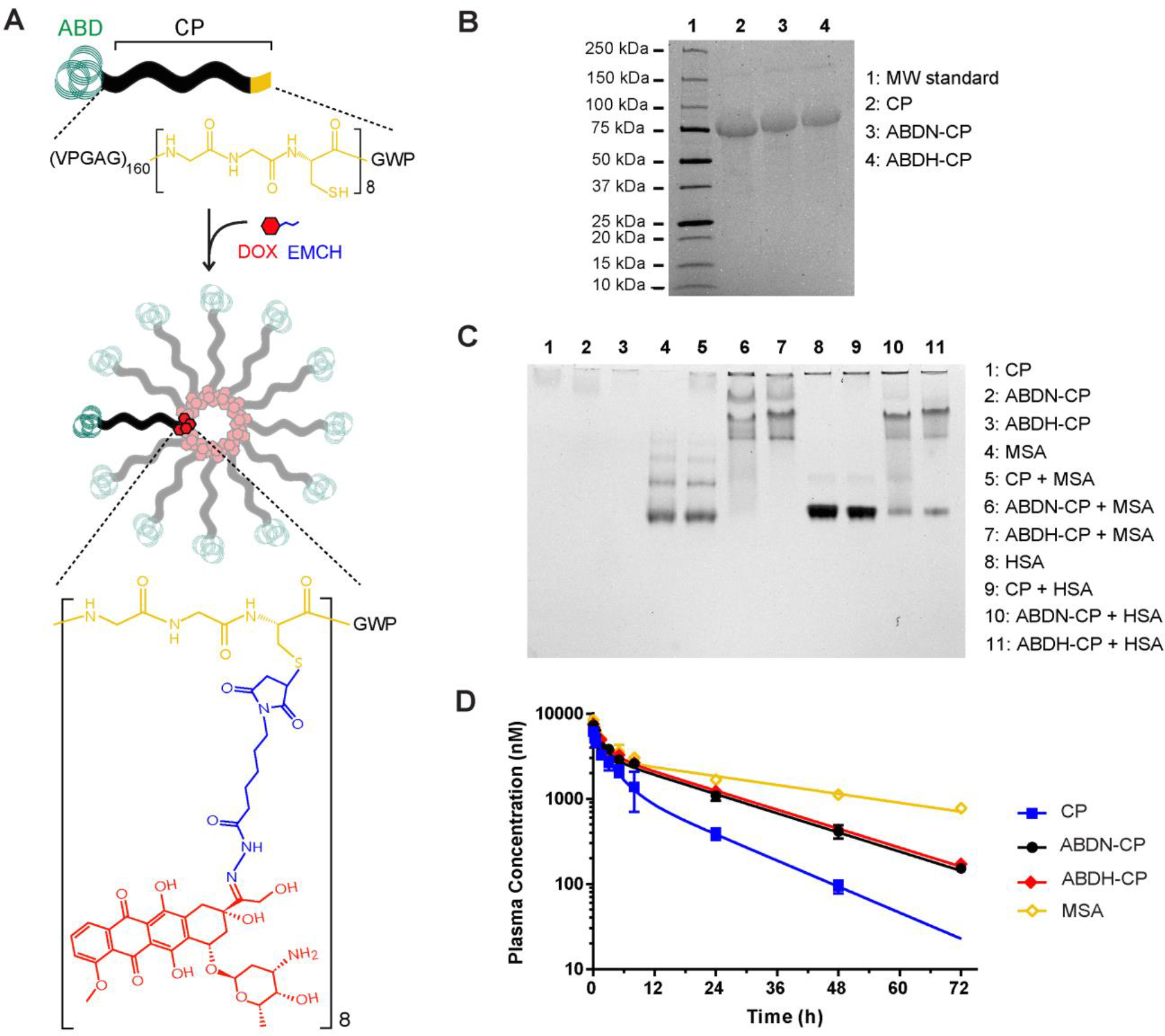
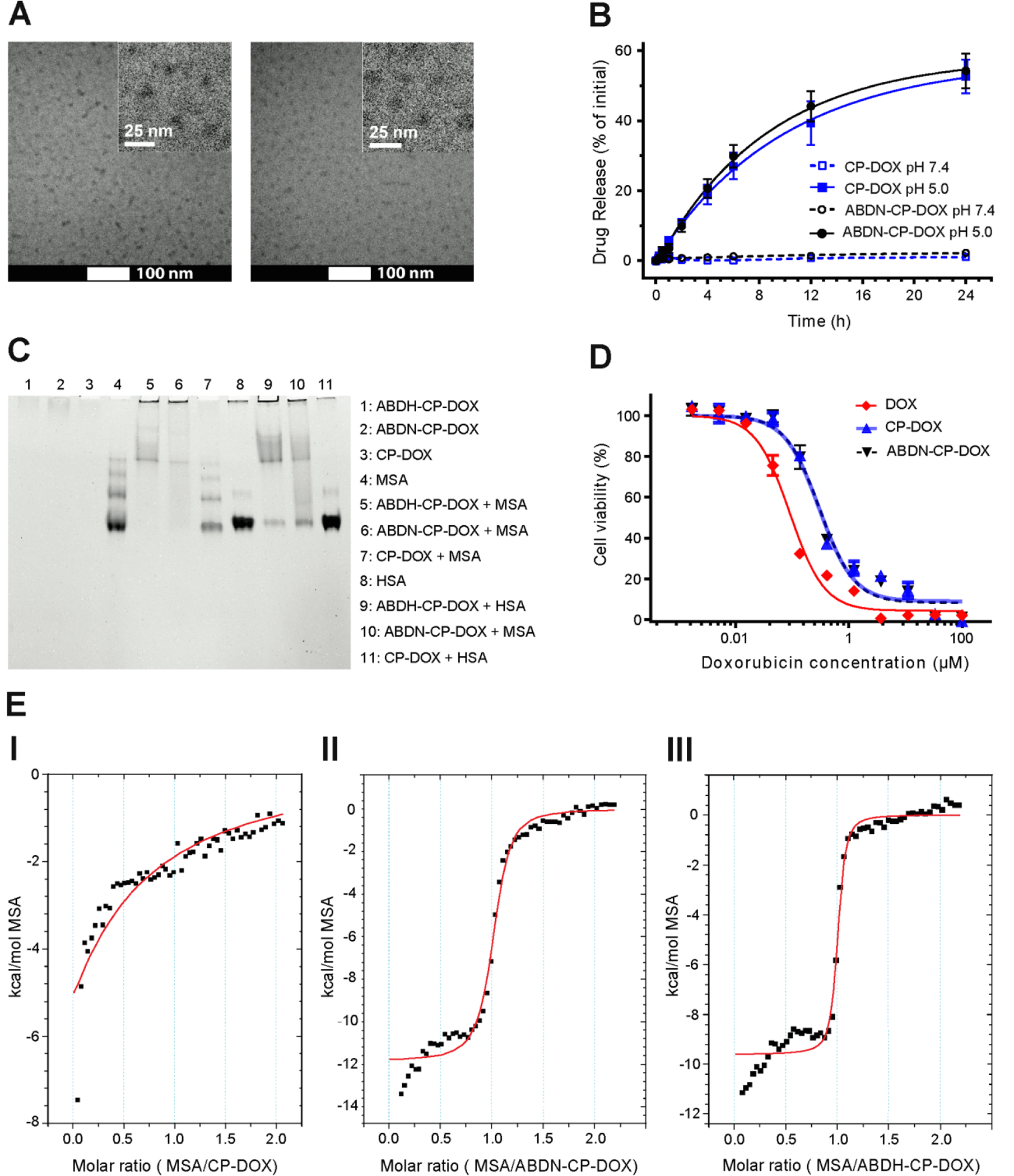
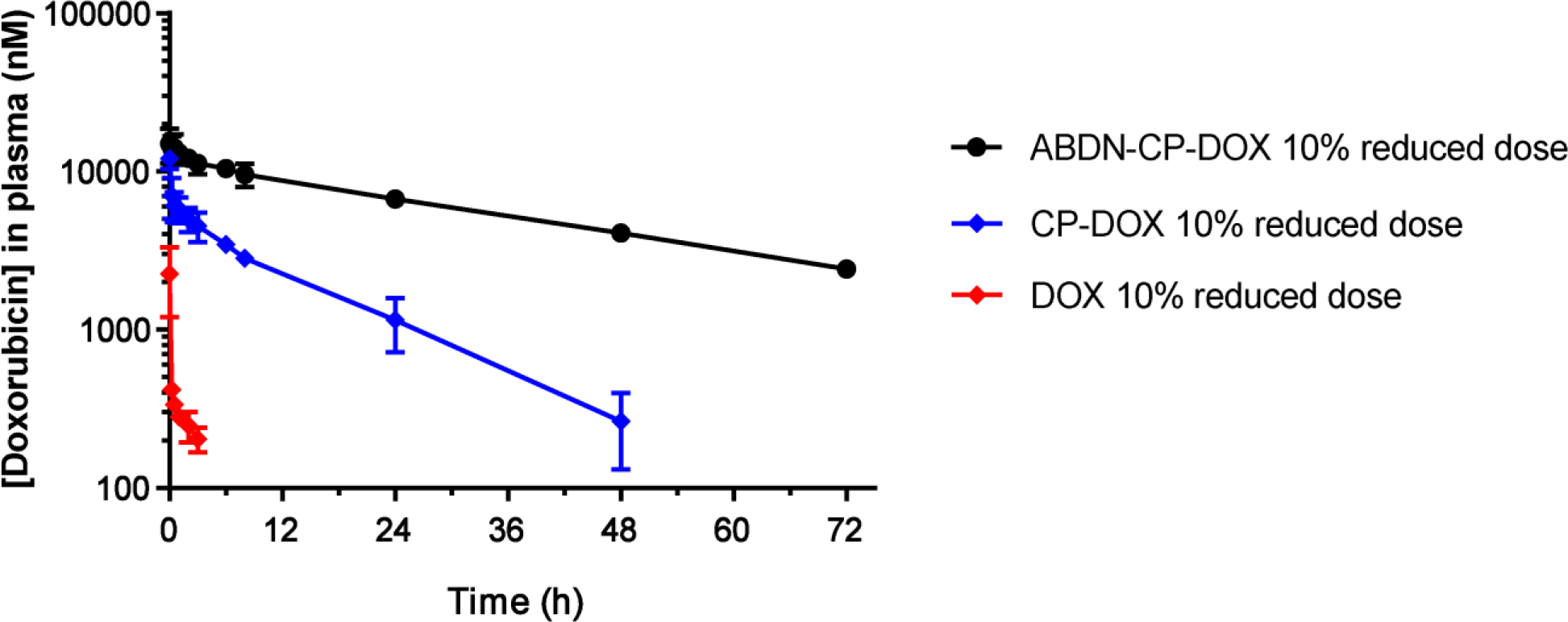
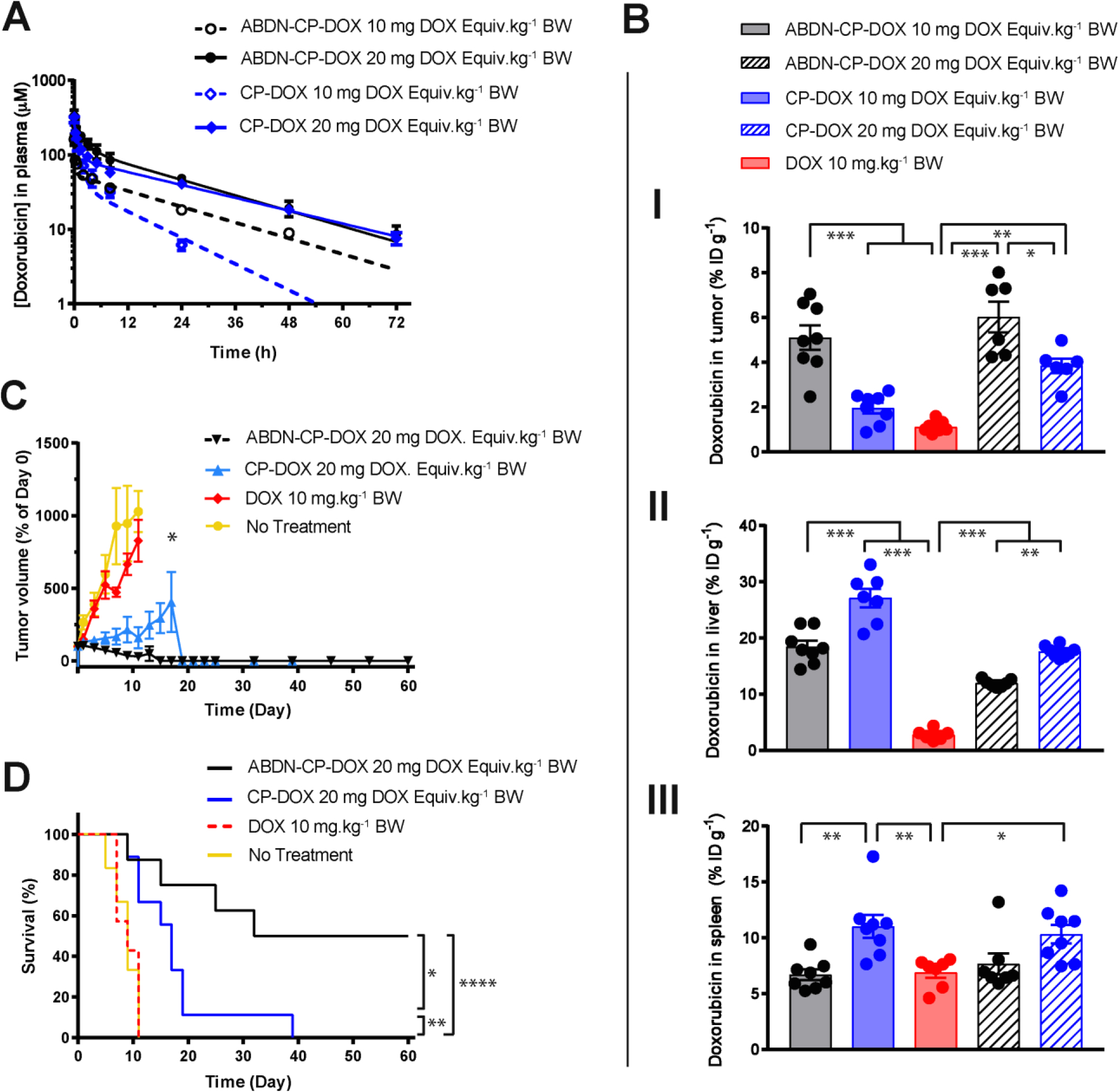
We report the development of drug-encapsulating nanoparticles that bind endogenous albumin upon intravenous injection and evaluate their in vivo performance in a murine as well as canine animal model. The gene encoding a protein-G derived albumin binding domain (ABD) was fused to that of a chimeric polypeptide (CP), and the ABD-CP fusion was recombinantly synthesized by bacterial expression of the gene. Doxorubicin (DOX) was conjugated to the C-terminus of the ABD-CP fusion, and conjugation of multiple copies of the drug to one end of the ABD-CP triggered its self-assembly into ∼100 nm diameter spherical micelles. ABD-decorated micelles exhibited submicromolar binding affinity for albumin and also preserved their spherical morphology in the presence of albumin. In a murine model, albumin-binding micelles exhibited dose-independent pharmacokinetics, whereas naked micelles exhibited dose-dependent pharmacokinetics. In addition, in a canine model, albumin binding micelles resulted in a 3-fold increase in plasma half-life and 6-fold increase in plasma exposure as defined by the area under the curve (AUC) of the drug, compared with naked micelles. Furthermore, in a murine colon carcinoma model, albumin-binding nanoparticles demonstrated lower uptake by the reticuloendothelial system (RES) system organs, the liver and spleen, that are the main target organs of toxicity for nanoparticulate delivery systems and higher uptake by the tumor than naked micelles. The increased uptake by s.c. C26 colon carcinoma tumors in mice translated to a wider therapeutic window of doses ranging from 20 to 60 mg equivalent of DOX per kg body weight (mg DOX equiv·kg-1 BW) for albumin-binding ABD-CP-DOX micelles, as compared to naked micelles that were only effective at their maximum tolerated dose of 40 mg DOX equiv·kg-1 BW.
Keywords: Micelles; cancer; canine model; drug delivery; elastin like polypeptide; endogenous albumin.
Conflict of interest statement
Conflicts of Interest
A.C. serves as a scientific advisor and board member for PhaseBio Pharmaceuticals, Inc., which has licensed the ELP technology for drug delivery from Duke University. The other authors declare no competing financial interest.
Figures
Similar articles
-
Targeted delivery of Doxorubicin by folic acid-decorated dual functional nanocarrier.Mol Pharm. 2014 Nov 3;11(11):4164-78. doi: 10.1021/mp500389v. Epub 2014 Sep 29. Mol Pharm. 2014. PMID: 25265550 Free PMC article.
-
Extending Half Life of H-Ferritin Nanoparticle by Fusing Albumin Binding Domain for Doxorubicin Encapsulation.Biomacromolecules. 2018 Mar 12;19(3):773-781. doi: 10.1021/acs.biomac.7b01545. Epub 2018 Feb 5. Biomacromolecules. 2018. PMID: 29328653
-
Conjugate of Doxorubicin to Albumin-Binding Peptide Outperforms Aldoxorubicin.Small. 2019 Mar;15(12):e1804452. doi: 10.1002/smll.201804452. Epub 2019 Feb 13. Small. 2019. PMID: 30756483 Free PMC article.
-
Starch-based "smart" nanomicelles: Potential delivery systems for doxorubicin.Drug Dev Res. 2024 Sep;85(6):e22253. doi: 10.1002/ddr.22253. Drug Dev Res. 2024. PMID: 39207174 Review.
-
Role of Albumin as a Targeted Drug Carrier in the Management of Rheumatoid Arthritis: A Comprehensive Review.Mol Pharm. 2023 Nov 6;20(11):5345-5358. doi: 10.1021/acs.molpharmaceut.3c00581. Epub 2023 Oct 23. Mol Pharm. 2023. PMID: 37870420 Review.
Cited by
-
Supramolecular nanomedicines through rational design of self-assembling prodrugs.Trends Pharmacol Sci. 2022 Jun;43(6):510-521. doi: 10.1016/j.tips.2022.03.003. Epub 2022 Apr 19. Trends Pharmacol Sci. 2022. PMID: 35459589 Free PMC article. Review.
-
Composites Additive Manufacturing for Space Applications: A Review.Materials (Basel). 2022 Jul 5;15(13):4709. doi: 10.3390/ma15134709. Materials (Basel). 2022. PMID: 35806833 Free PMC article. Review.
-
Extracellular vesicles engineered to bind albumin demonstrate extended circulation time and lymph node accumulation in mouse models.J Extracell Vesicles. 2022 Jul;11(7):e12248. doi: 10.1002/jev2.12248. J Extracell Vesicles. 2022. PMID: 35879268 Free PMC article.
-
Novel fusion peptides deliver exosomes to modify injectable thermo-sensitive hydrogels for bone regeneration.Mater Today Bio. 2021 Dec 27;13:100195. doi: 10.1016/j.mtbio.2021.100195. eCollection 2022 Jan. Mater Today Bio. 2021. PMID: 35024598 Free PMC article.
-
A Nanoparticle's Journey to the Tumor: Strategies to Overcome First-Pass Metabolism and Their Limitations.Cancers (Basel). 2022 Mar 29;14(7):1741. doi: 10.3390/cancers14071741. Cancers (Basel). 2022. PMID: 35406513 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
