

Folding pathway of an Ig domain is conserved on and off the ribosome
- PMID: 30413621
- PMCID: PMC6275497
- DOI: 10.1073/pnas.1810523115
Folding pathway of an Ig domain is conserved on and off the ribosome
Abstract
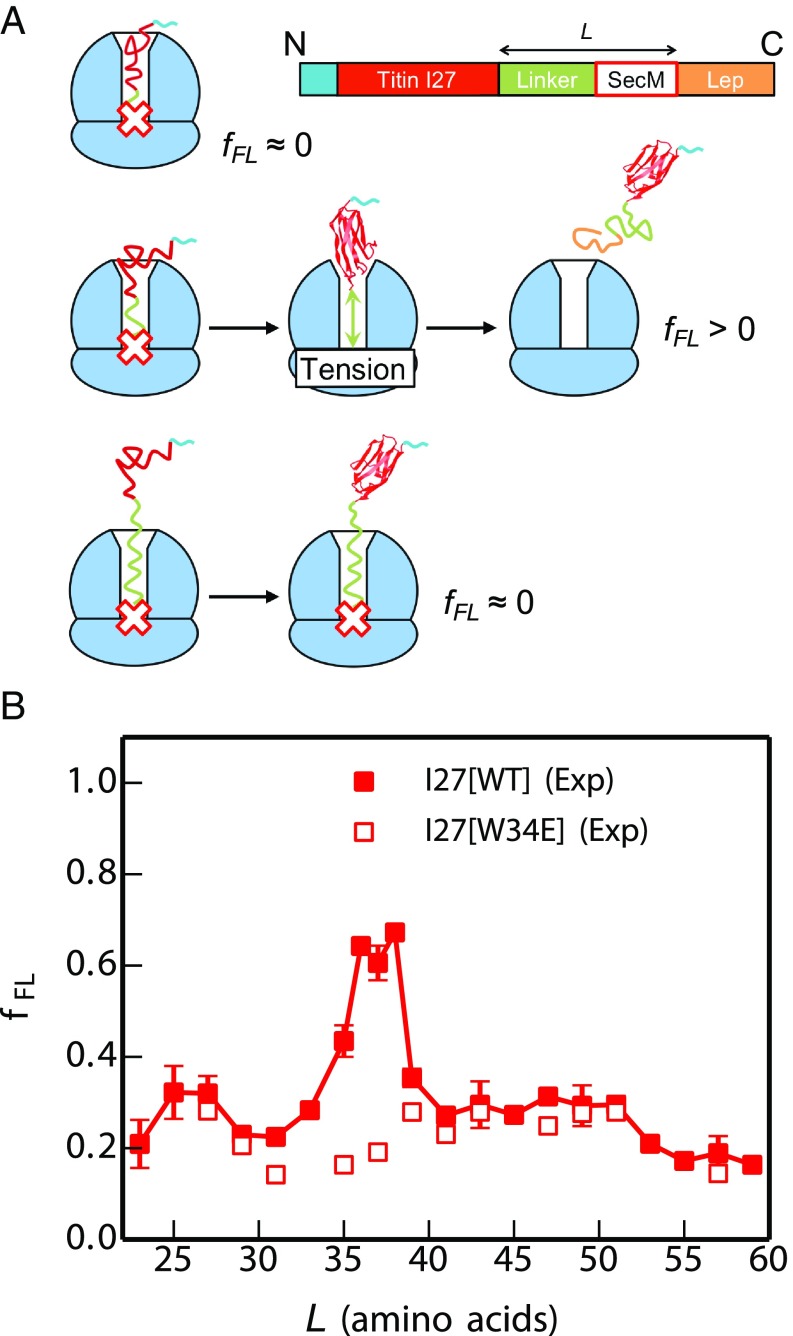
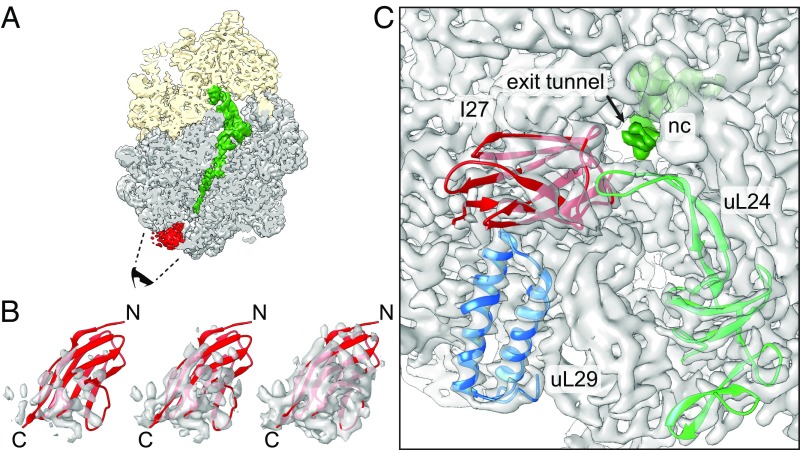
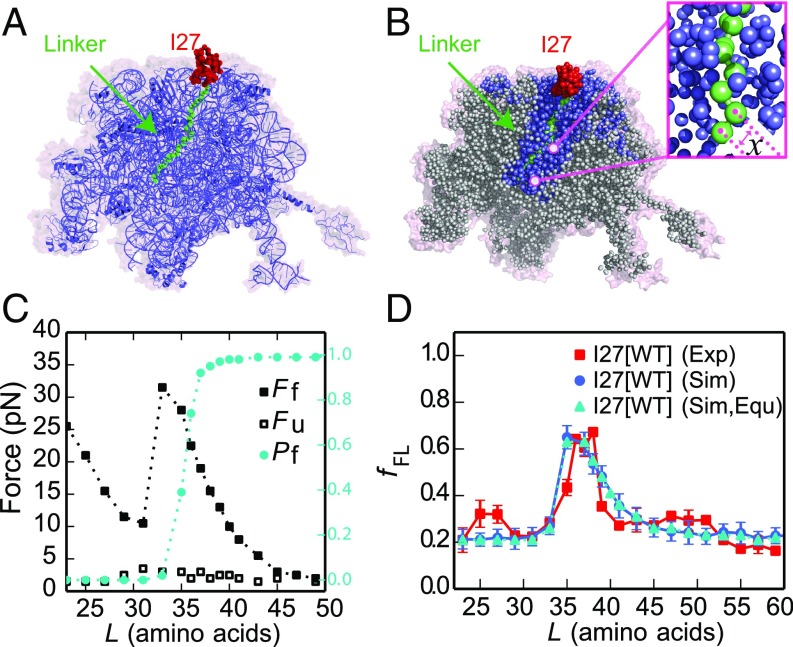
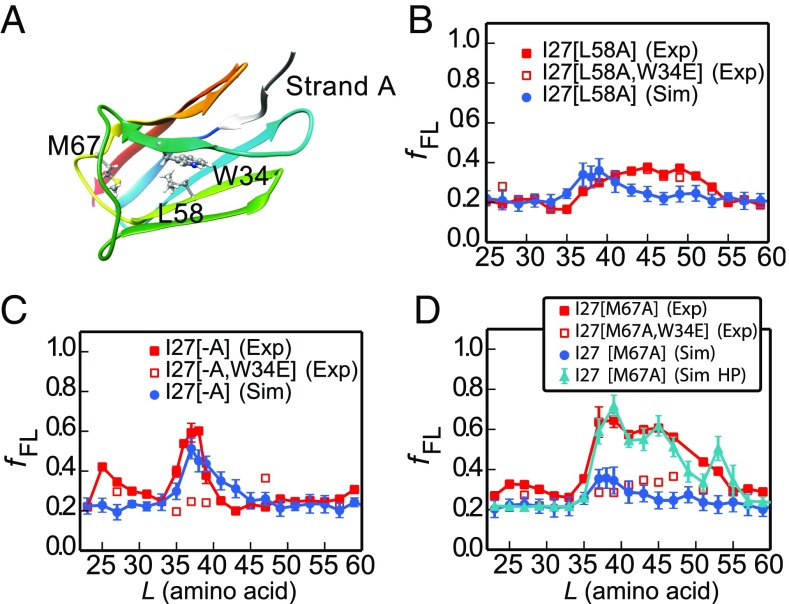
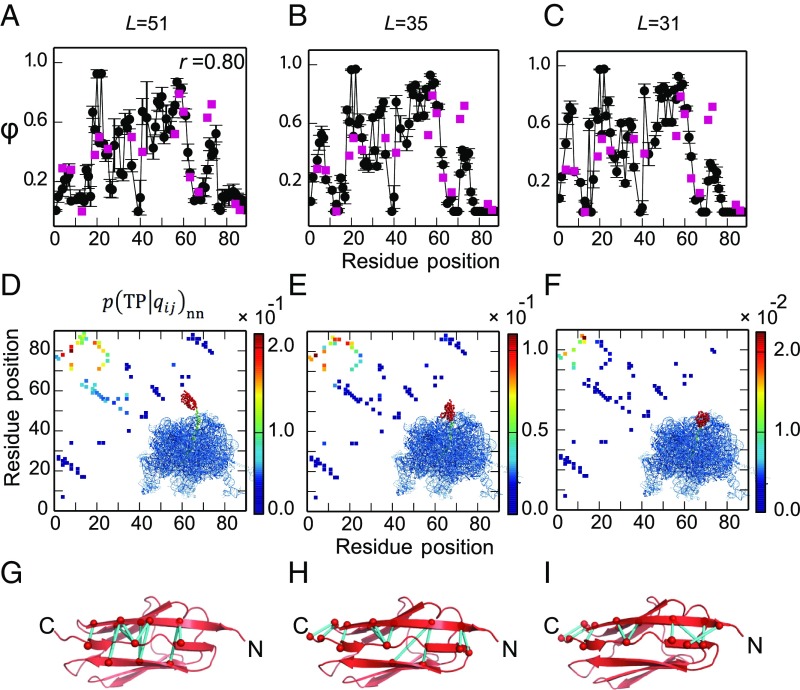
Proteins that fold cotranslationally may do so in a restricted configurational space, due to the volume occupied by the ribosome. How does this environment, coupled with the close proximity of the ribosome, affect the folding pathway of a protein? Previous studies have shown that the cotranslational folding process for many proteins, including small, single domains, is directly affected by the ribosome. Here, we investigate the cotranslational folding of an all-β Ig domain, titin I27. Using an arrest peptide-based assay and structural studies by cryo-EM, we show that I27 folds in the mouth of the ribosome exit tunnel. Simulations that use a kinetic model for the force dependence of escape from arrest accurately predict the fraction of folded protein as a function of length. We used these simulations to probe the folding pathway on and off the ribosome. Our simulations-which also reproduce experiments on mutant forms of I27-show that I27 folds, while still sequestered in the mouth of the ribosome exit tunnel, by essentially the same pathway as free I27, with only subtle shifts of critical contacts from the C to the N terminus.
Keywords: arrest peptide; fraction folded; kinetic model; mechanical force; molecular simulation.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
A small single-domain protein folds through the same pathway on and off the ribosome.Proc Natl Acad Sci U S A. 2018 Nov 27;115(48):12206-12211. doi: 10.1073/pnas.1810517115. Epub 2018 Nov 8. Proc Natl Acad Sci U S A. 2018. PMID: 30409803 Free PMC article.
-
The shape of the bacterial ribosome exit tunnel affects cotranslational protein folding.Elife. 2018 Nov 26;7:e36326. doi: 10.7554/eLife.36326. Elife. 2018. PMID: 30475203 Free PMC article.
-
Influence of Secondary-Structure Folding on the Mutually Exclusive Folding Process of GL5/I27 Protein: Evidence from Molecular Dynamics Simulations.Int J Mol Sci. 2016 Nov 23;17(11):1962. doi: 10.3390/ijms17111962. Int J Mol Sci. 2016. PMID: 27886109 Free PMC article.
-
Cotranslational Folding of Proteins on the Ribosome.Biomolecules. 2020 Jan 7;10(1):97. doi: 10.3390/biom10010097. Biomolecules. 2020. PMID: 31936054 Free PMC article. Review.
-
The ribosome and its role in protein folding: looking through a magnifying glass.Acta Crystallogr D Struct Biol. 2017 Jun 1;73(Pt 6):509-521. doi: 10.1107/S2059798317007446. Epub 2017 May 31. Acta Crystallogr D Struct Biol. 2017. PMID: 28580913 Free PMC article. Review.
Cited by
-
Force transduction creates long-ranged coupling in ribosomes stalled by arrest peptides.Biophys J. 2021 Jun 15;120(12):2425-2435. doi: 10.1016/j.bpj.2021.03.041. Epub 2021 Apr 29. Biophys J. 2021. PMID: 33932440 Free PMC article.
-
Hydrophobic and electrostatic interactions modulate protein escape at the ribosomal exit tunnel.Biophys J. 2021 Nov 2;120(21):4798-4808. doi: 10.1016/j.bpj.2021.09.027. Epub 2021 Sep 21. Biophys J. 2021. PMID: 34555360 Free PMC article.
-
Cotranslational folding cooperativity of contiguous domains of α-spectrin.Proc Natl Acad Sci U S A. 2020 Jun 23;117(25):14119-14126. doi: 10.1073/pnas.1909683117. Epub 2020 Jun 8. Proc Natl Acad Sci U S A. 2020. PMID: 32513720 Free PMC article.
-
Mechanisms of Cotranslational Protein Maturation in Bacteria.Front Mol Biosci. 2021 May 25;8:689755. doi: 10.3389/fmolb.2021.689755. eCollection 2021. Front Mol Biosci. 2021. PMID: 34113653 Free PMC article. Review.
-
Synthesis runs counter to directional folding of a nascent protein domain.Nat Commun. 2020 Oct 9;11(1):5096. doi: 10.1038/s41467-020-18921-8. Nat Commun. 2020. PMID: 33037221 Free PMC article.
References
-
- Nilsson OB, et al. Cotranslational folding of spectrin domains via partially structured states. Nat Struct Mol Biol. 2017;24:221–225. - PubMed
-
- Nilsson OB, Müller-Lucks A, Kramer G, Bukau B, von Heijne G. Trigger factor reduces the force exerted on the nascent chain by a cotranslationally folding protein. J Mol Biol. 2016;428:1356–1364. - PubMed
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. - PubMed
-
- Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: The energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
