

Soluble CD74 Reroutes MIF/CXCR4/AKT-Mediated Survival of Cardiac Myofibroblasts to Necroptosis
- PMID: 30371153
- PMCID: PMC6201423
- DOI: 10.1161/JAHA.118.009384
Soluble CD74 Reroutes MIF/CXCR4/AKT-Mediated Survival of Cardiac Myofibroblasts to Necroptosis
Abstract
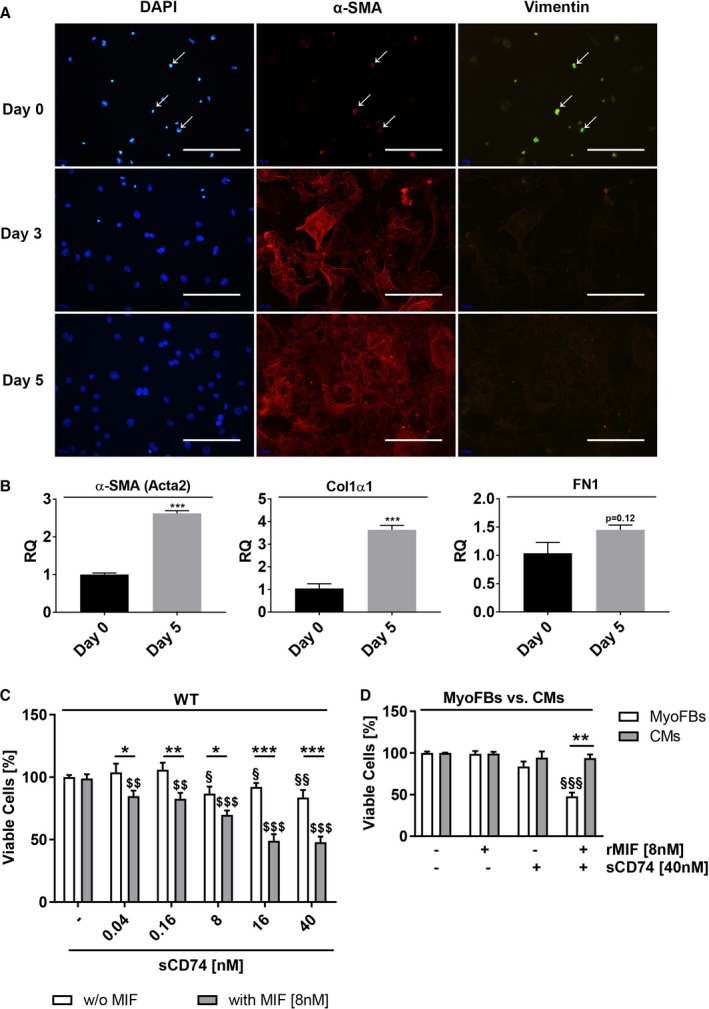
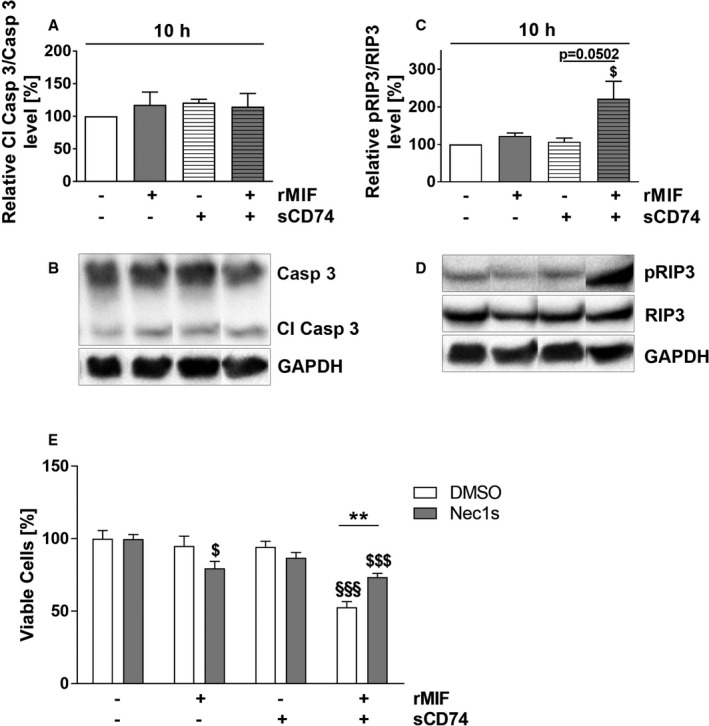
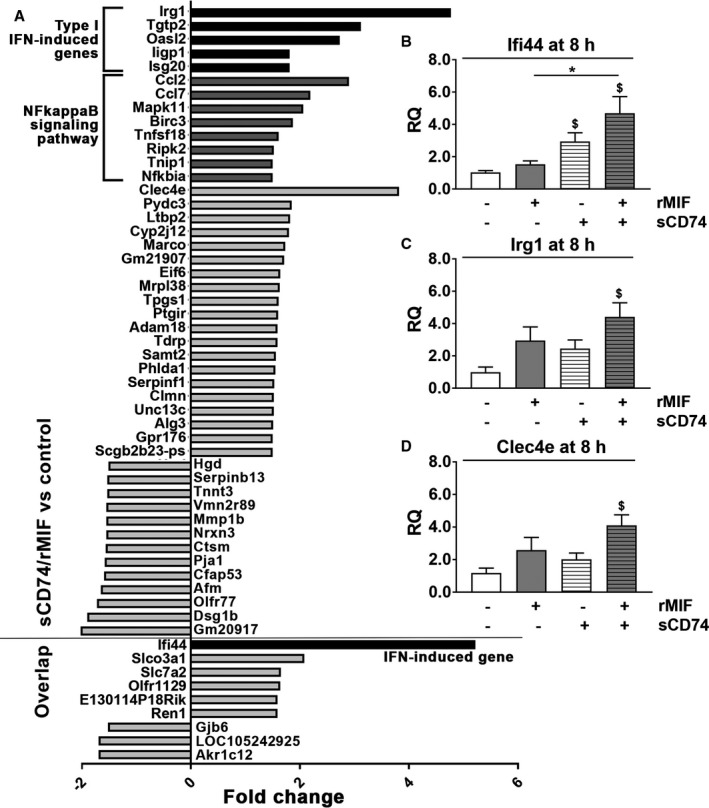
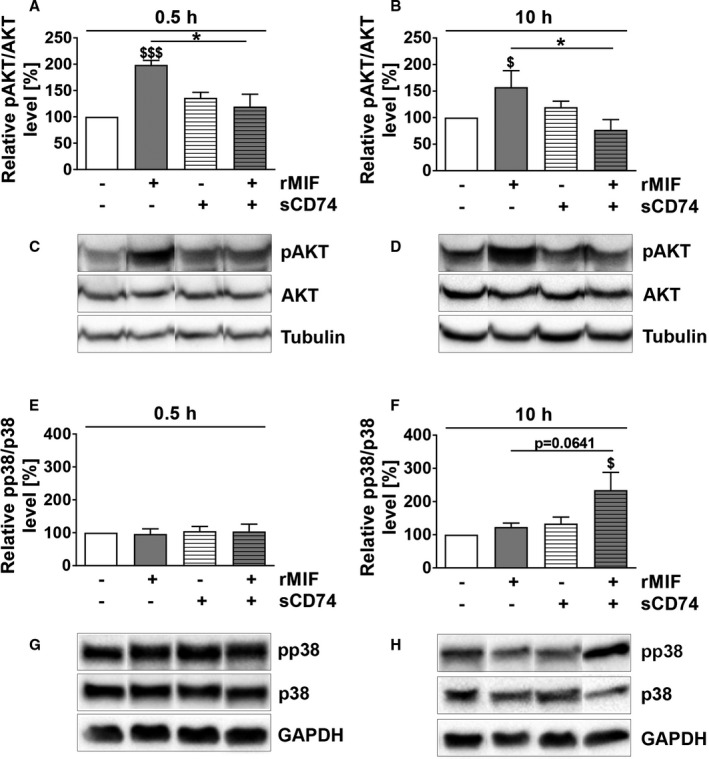
Background Although macrophage migration inhibitory factor ( MIF ) has been demonstrated to mediate cardioprotection in ischemia/reperfusion injury and antagonize fibrotic effects through its receptor, CD 74, the function of the soluble CD 74 receptor ectodomain ( sCD 74) and its interaction with circulating MIF have not been explored in cardiac disease. Methods and Results Cardiac fibroblasts were isolated from hearts of neonatal mice and differentiated into myofibroblasts. Co-treatment with recombinant MIF and sCD 74 induced cell death ( P<0.001), which was mediated by receptor-interacting serine/threonine-protein kinase ( RIP) 1/ RIP 3-dependent necroptosis ( P=0.0376). This effect was specific for cardiac fibroblasts and did not affect cardiomyocytes. Gene expression analyses using microarray and RT - qPCR technology revealed a 4-fold upregulation of several interferon-induced genes upon co-treatment of myofibroblasts with sCD 74 and MIF (Ifi44: P=0.011; Irg1: P=0.022; Clec4e: P=0.011). Furthermore, Western blot analysis confirmed the role of sCD 74 as a modulator of MIF signaling by diminishing MIF -mediated protein kinase B ( AKT) activation ( P=0.0197) and triggering p38 activation ( P=0.0641). We obtained evidence that sCD 74 inhibits MIF -mediated survival pathway through the C-X-C chemokine receptor 4/ AKT axis, enabling the induction of CD 74-dependent necroptotic processes in cardiac myofibroblasts. Preliminary clinical data revealed a lowered sCD 74/ MIF ratio in heart failure patients (17.47±10.09 versus 1.413±0.6244). Conclusions These findings suggest that treatment of cardiac myofibroblasts with sCD 74 and MIF induces necroptosis, offering new insights into the mechanism of myofibroblast depletion during scar maturation. Preliminary clinical data provided first evidence about a clinical relevance of the sCD 74/ MIF axis in heart failure, suggesting that these proteins may be a promising target to modulate cardiac remodeling and disease progression in heart failure.
Keywords: cell death; heart failure; macrophage migration inhibitory factor; myocardial fibrosis; myofibroblast; necroptosis; soluble CD74.
Figures
Similar articles
-
Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74.Cell Signal. 2011 Jan;23(1):135-44. doi: 10.1016/j.cellsig.2010.08.013. Epub 2010 Aug 31. Cell Signal. 2011. PMID: 20807568 Free PMC article.
-
Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity.Oncogene. 2007 Aug 2;26(35):5046-59. doi: 10.1038/sj.onc.1210318. Epub 2007 Feb 19. Oncogene. 2007. PMID: 17310986
-
Macrophage migration inhibitory factor promotes cardiac stem cell proliferation and endothelial differentiation through the activation of the PI3K/Akt/mTOR and AMPK pathways.Int J Mol Med. 2016 May;37(5):1299-309. doi: 10.3892/ijmm.2016.2542. Epub 2016 Mar 29. Int J Mol Med. 2016. PMID: 27035848 Free PMC article.
-
MIF, CD74 and other partners in kidney disease: tales of a promiscuous couple.Cytokine Growth Factor Rev. 2013 Feb;24(1):23-40. doi: 10.1016/j.cytogfr.2012.08.001. Epub 2012 Sep 7. Cytokine Growth Factor Rev. 2013. PMID: 22959722 Review.
-
Evolving complexity of MIF signaling.Cell Signal. 2019 May;57:76-88. doi: 10.1016/j.cellsig.2019.01.006. Epub 2019 Jan 23. Cell Signal. 2019. PMID: 30682543 Review.
Cited by
-
Tissue Inhibitor of Metalloproteinases-1 Interacts with CD74 to Promote AKT Signaling, Monocyte Recruitment Responses, and Vascular Smooth Muscle Cell Proliferation.Cells. 2023 Jul 20;12(14):1899. doi: 10.3390/cells12141899. Cells. 2023. PMID: 37508563 Free PMC article.
-
D-dopachrome tautomerase from Japanese sea bass ( Lateolabrax japonicus) is a chemokine-like cytokine and functional homolog of macrophage migration inhibitory factor.Zool Res. 2020 Jan 18;41(1):39-50. doi: 10.24272/j.issn.2095-8137.2020.003. Zool Res. 2020. PMID: 31709785 Free PMC article.
-
M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer.Biomater Res. 2022 Apr 11;26(1):14. doi: 10.1186/s40824-022-00260-y. Biomater Res. 2022. PMID: 35410296 Free PMC article.
-
Surfactant Protein-C Regulates Alveolar Type 2 Epithelial Cell Lineages via the CD74 Receptor.J Respir Biol Transl Med. 2024 Dec;1(4):10017. doi: 10.70322/jrbtm.2024.10017. Epub 2024 Oct 11. J Respir Biol Transl Med. 2024. PMID: 39553282 Free PMC article.
-
Targeting necroptosis as therapeutic potential in chronic myocardial infarction.J Biomed Sci. 2021 Apr 9;28(1):25. doi: 10.1186/s12929-021-00722-w. J Biomed Sci. 2021. PMID: 33836761 Free PMC article. Review.
References
-
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive summary: heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:447–454. - PubMed
-
- Lloyd‐Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel‐Smoller S, Wong ND, Wylie‐Rosett J. Executive summary: heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. - PubMed
-
- Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–1939. - PubMed
-
- Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
