

AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A
- PMID: 30262884
- PMCID: PMC6160451
- DOI: 10.1038/s41467-018-06172-7
AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A
Abstract
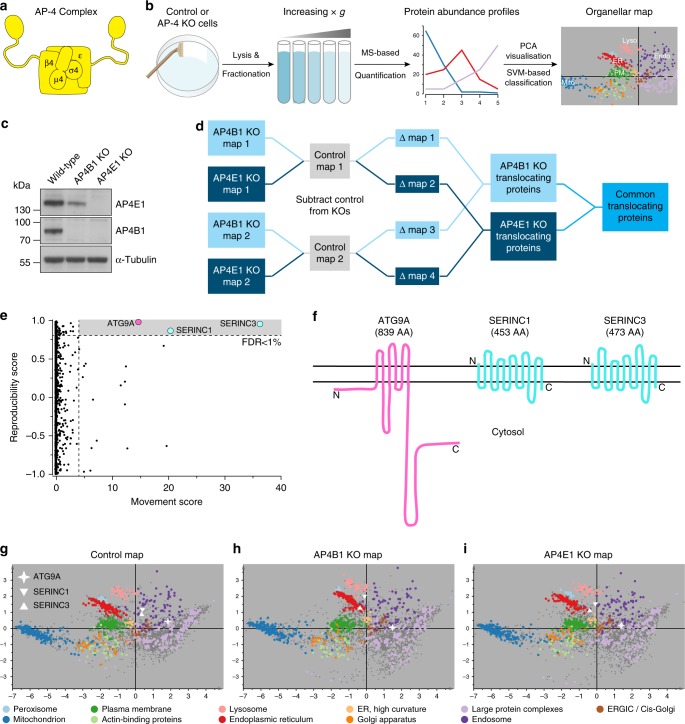
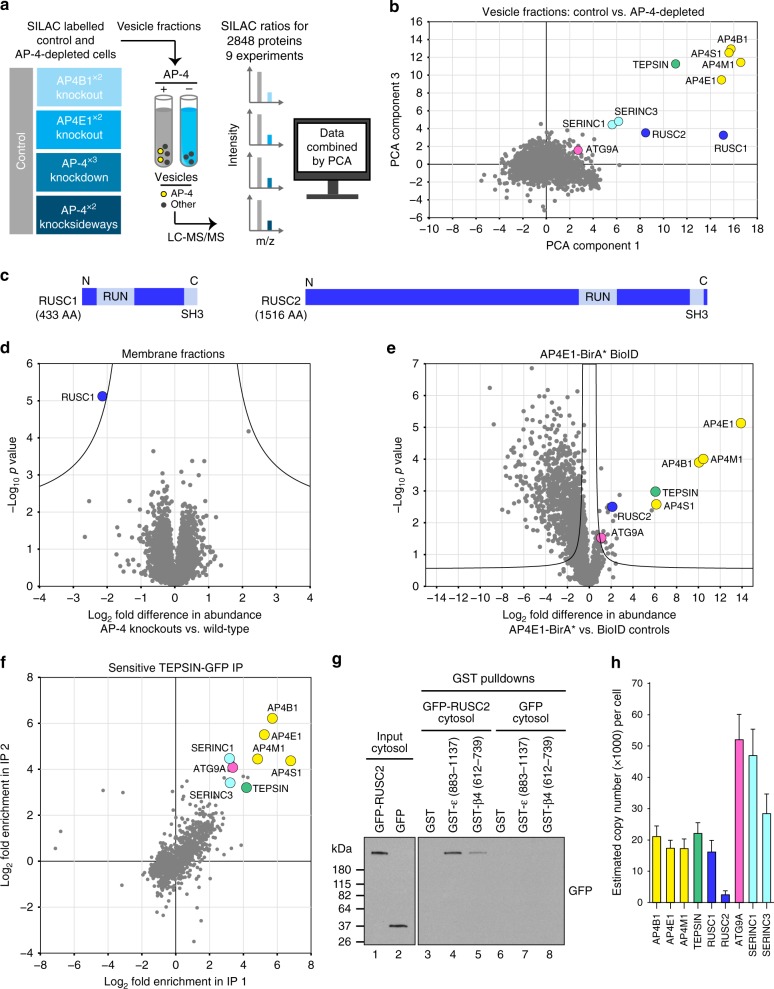
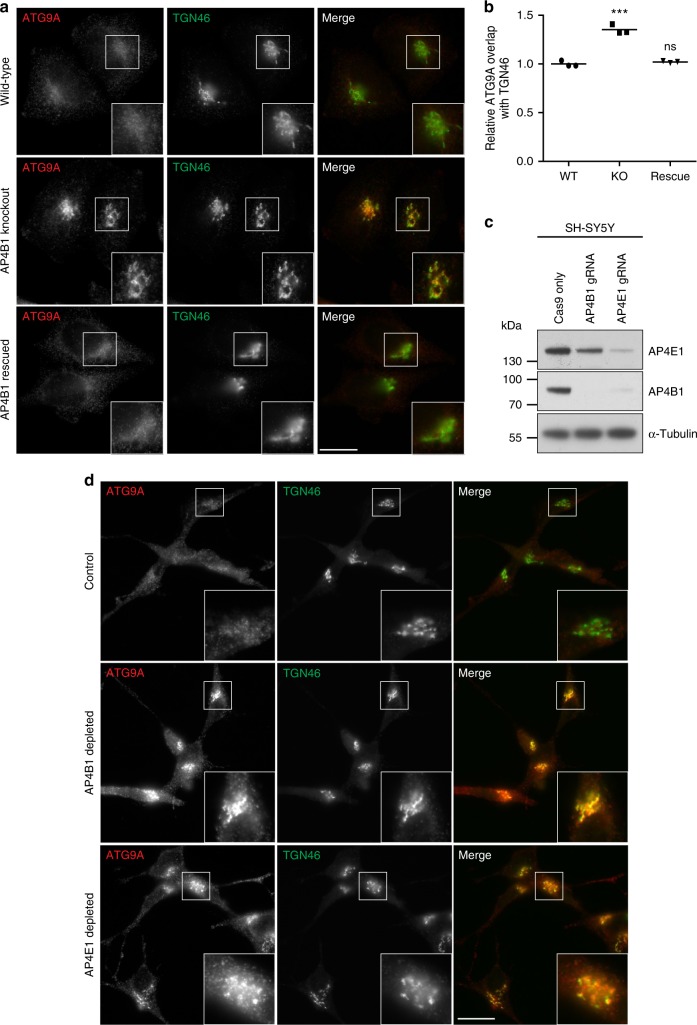
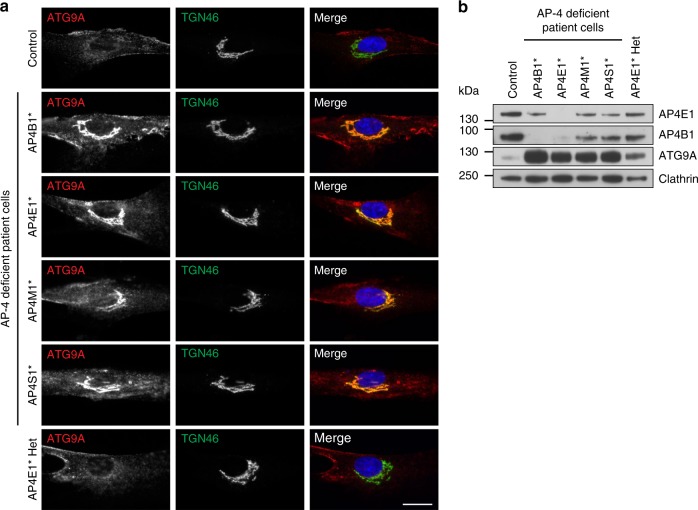
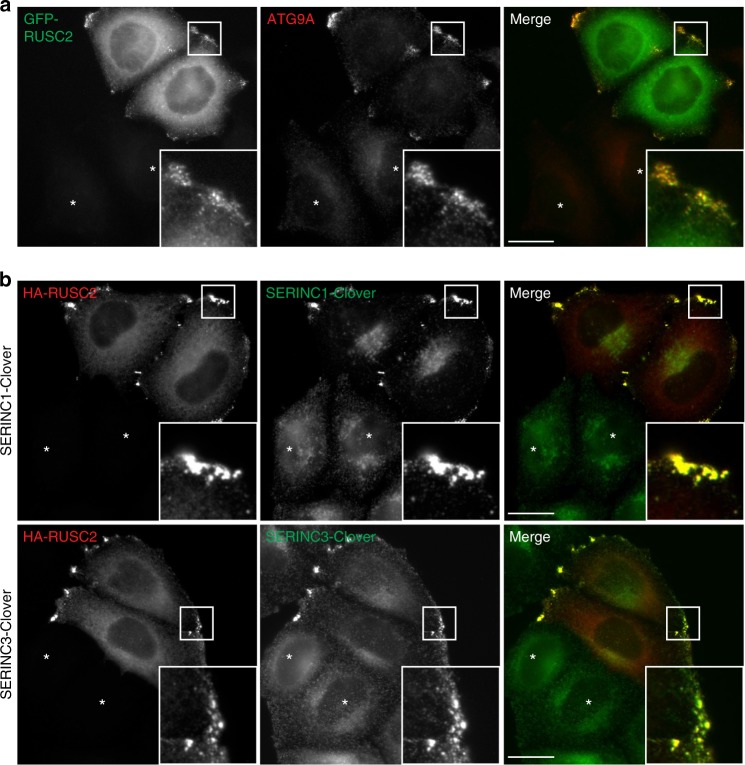
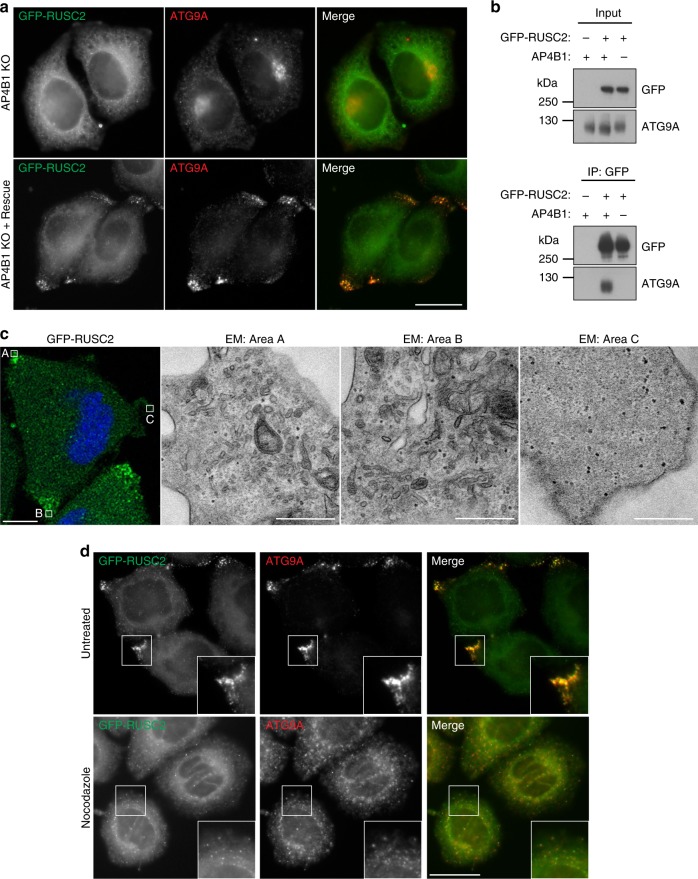
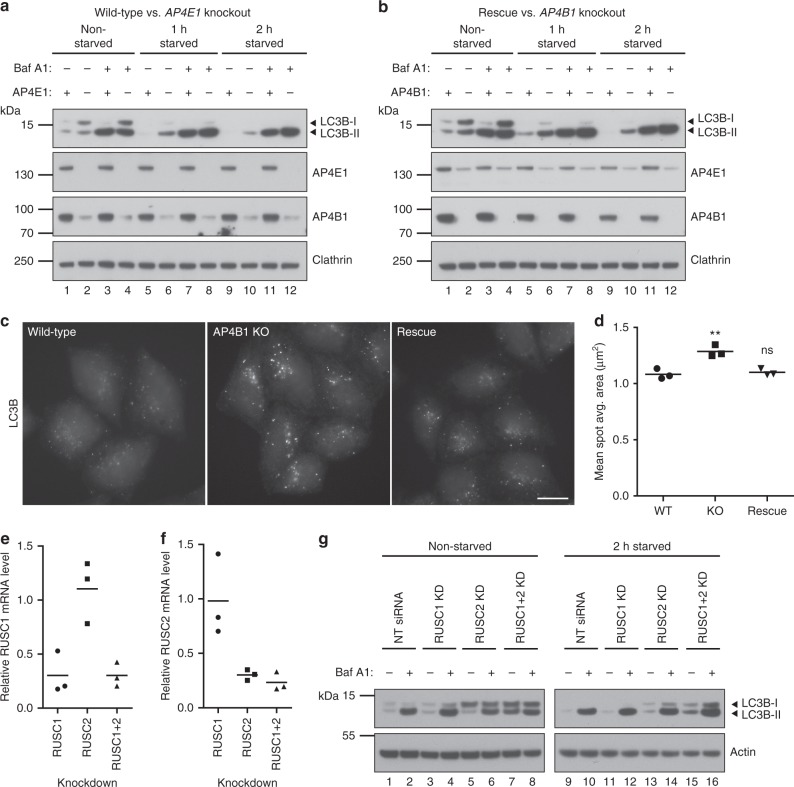
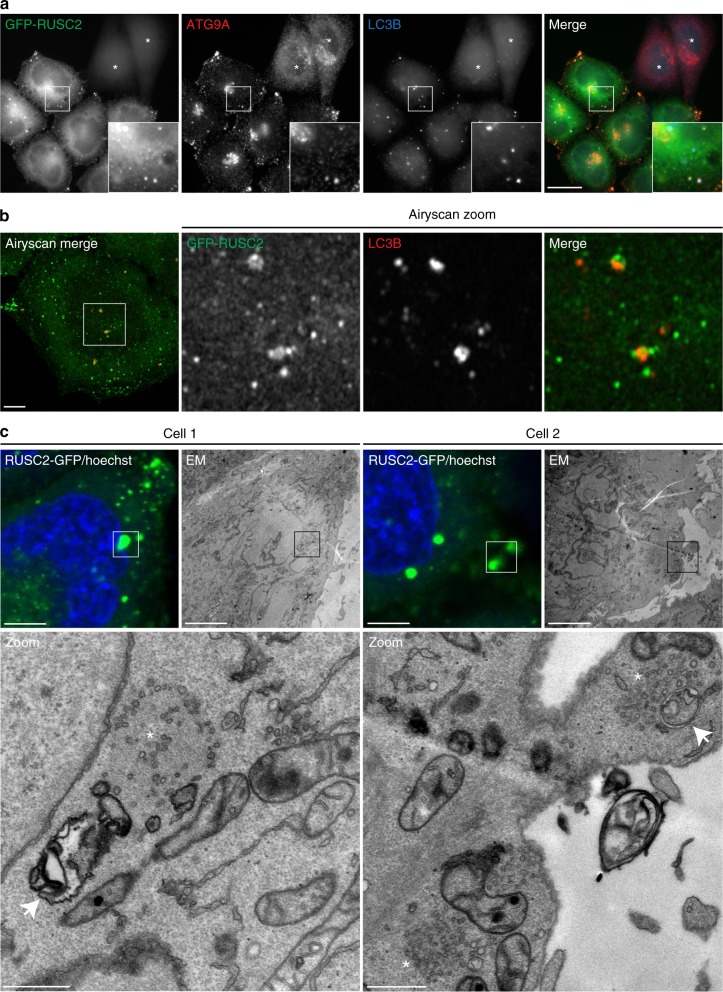
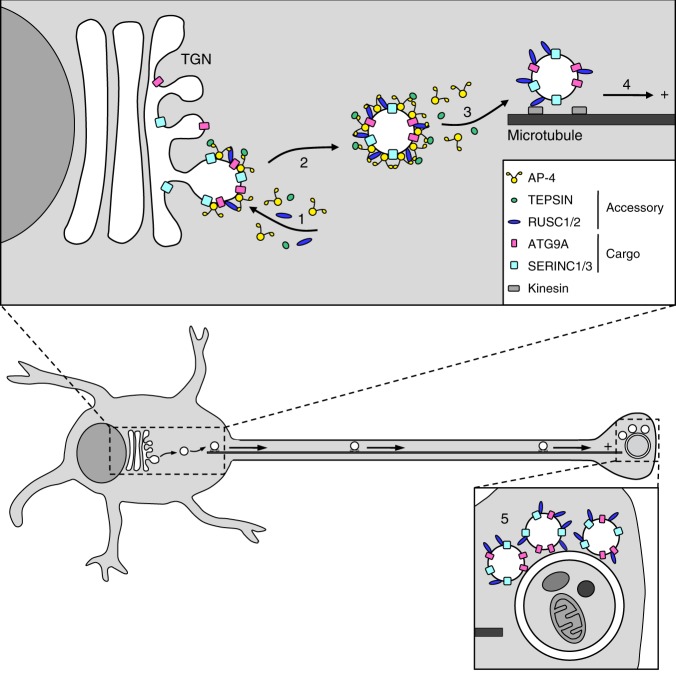
Adaptor protein 4 (AP-4) is an ancient membrane trafficking complex, whose function has largely remained elusive. In humans, AP-4 deficiency causes a severe neurological disorder of unknown aetiology. We apply unbiased proteomic methods, including 'Dynamic Organellar Maps', to find proteins whose subcellular localisation depends on AP-4. We identify three transmembrane cargo proteins, ATG9A, SERINC1 and SERINC3, and two AP-4 accessory proteins, RUSC1 and RUSC2. We demonstrate that AP-4 deficiency causes missorting of ATG9A in diverse cell types, including patient-derived cells, as well as dysregulation of autophagy. RUSC2 facilitates the transport of AP-4-derived, ATG9A-positive vesicles from the trans-Golgi network to the cell periphery. These vesicles cluster in close association with autophagosomes, suggesting they are the "ATG9A reservoir" required for autophagosome biogenesis. Our study uncovers ATG9A trafficking as a ubiquitous function of the AP-4 pathway. Furthermore, it provides a potential molecular pathomechanism of AP-4 deficiency, through dysregulated spatial control of autophagy.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Syntaxin 16's Newly Deciphered Roles in Autophagy.Cells. 2019 Dec 17;8(12):1655. doi: 10.3390/cells8121655. Cells. 2019. PMID: 31861136 Free PMC article. Review.
-
Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking.Hum Mol Genet. 2020 Jan 15;29(2):320-334. doi: 10.1093/hmg/ddz310. Hum Mol Genet. 2020. PMID: 31915823 Free PMC article.
-
The role of AP-4 in cargo export from the trans-Golgi network and hereditary spastic paraplegia.Biochem Soc Trans. 2020 Oct 30;48(5):1877-1888. doi: 10.1042/BST20190664. Biochem Soc Trans. 2020. PMID: 33084855 Review.
-
RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery.Mol Biol Cell. 2021 Nov 1;32(21):ar25. doi: 10.1091/mbc.E21-06-0295. Epub 2021 Aug 25. Mol Biol Cell. 2021. PMID: 34432492 Free PMC article.
-
The FTS-Hook-FHIP (FHF) complex interacts with AP-4 to mediate perinuclear distribution of AP-4 and its cargo ATG9A.Mol Biol Cell. 2020 Apr 15;31(9):963-979. doi: 10.1091/mbc.E19-11-0658. Epub 2020 Feb 19. Mol Biol Cell. 2020. PMID: 32073997 Free PMC article.
Cited by
-
Syntaxin 16's Newly Deciphered Roles in Autophagy.Cells. 2019 Dec 17;8(12):1655. doi: 10.3390/cells8121655. Cells. 2019. PMID: 31861136 Free PMC article. Review.
-
The core autophagy protein ATG9A controls dynamics of cell protrusions and directed migration.J Cell Biol. 2022 Mar 7;221(3):e202106014. doi: 10.1083/jcb.202106014. Epub 2022 Feb 18. J Cell Biol. 2022. PMID: 35180289 Free PMC article.
-
The different autophagy degradation pathways and neurodegeneration.Neuron. 2022 Mar 16;110(6):935-966. doi: 10.1016/j.neuron.2022.01.017. Epub 2022 Feb 7. Neuron. 2022. PMID: 35134347 Free PMC article. Review.
-
Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking.Hum Mol Genet. 2020 Jan 15;29(2):320-334. doi: 10.1093/hmg/ddz310. Hum Mol Genet. 2020. PMID: 31915823 Free PMC article.
-
Structural basis for ATG9A recruitment to the ULK1 complex in mitophagy initiation.Sci Adv. 2023 Feb 15;9(7):eadg2997. doi: 10.1126/sciadv.adg2997. Epub 2023 Feb 15. Sci Adv. 2023. PMID: 36791199 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
