

Structural principles of SNARE complex recognition by the AAA+ protein NSF
- PMID: 30198481
- PMCID: PMC6160233
- DOI: 10.7554/eLife.38888
Structural principles of SNARE complex recognition by the AAA+ protein NSF
Abstract
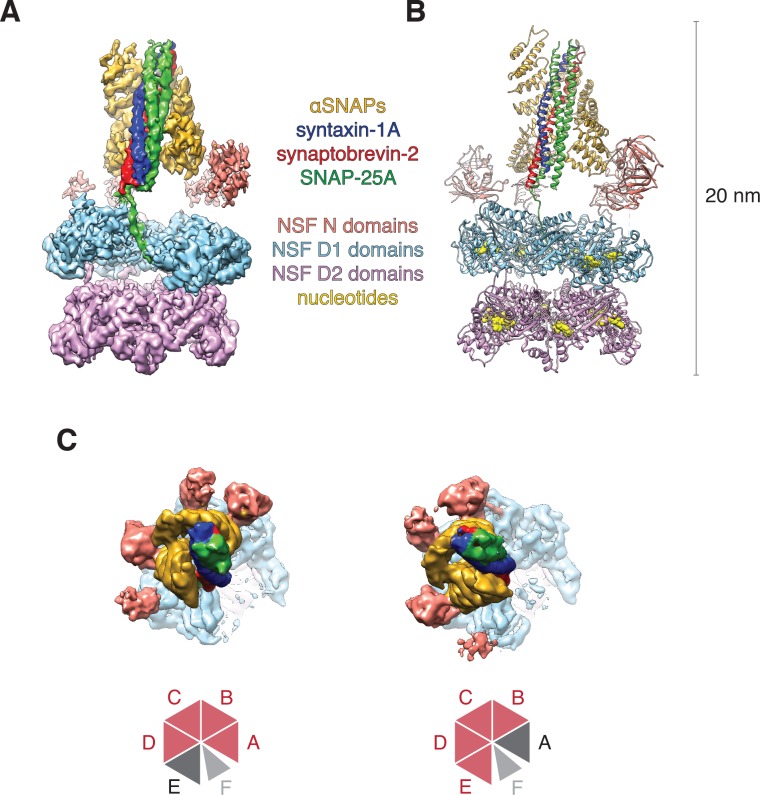
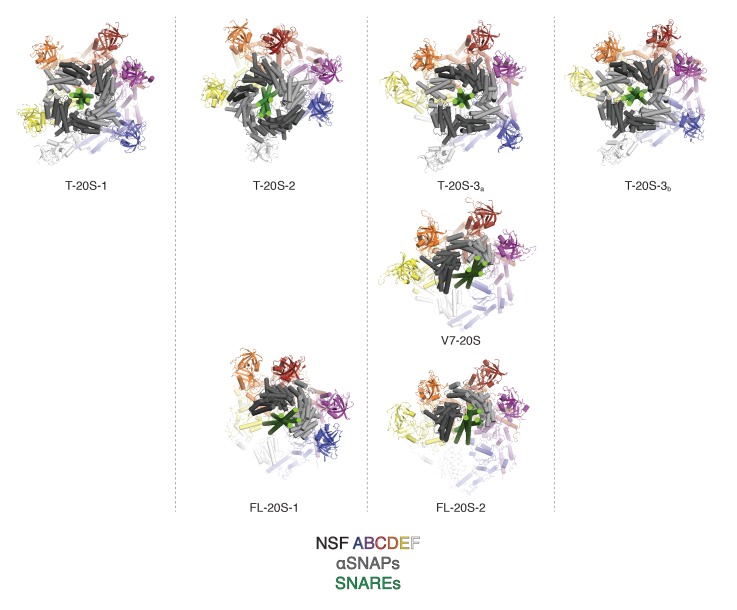
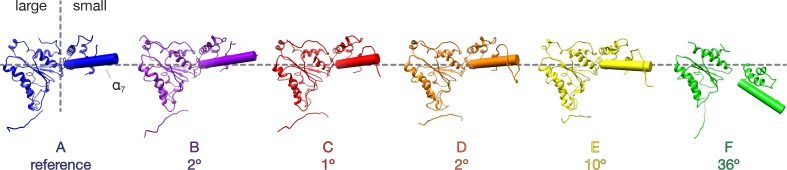
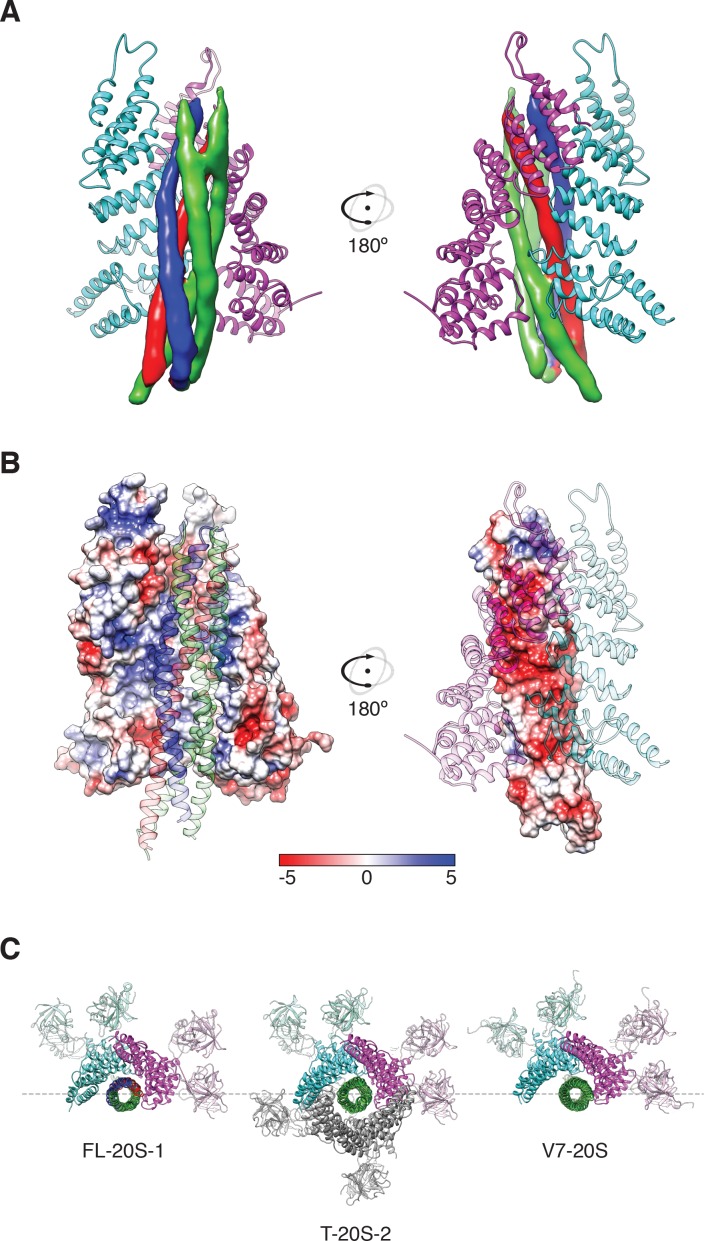
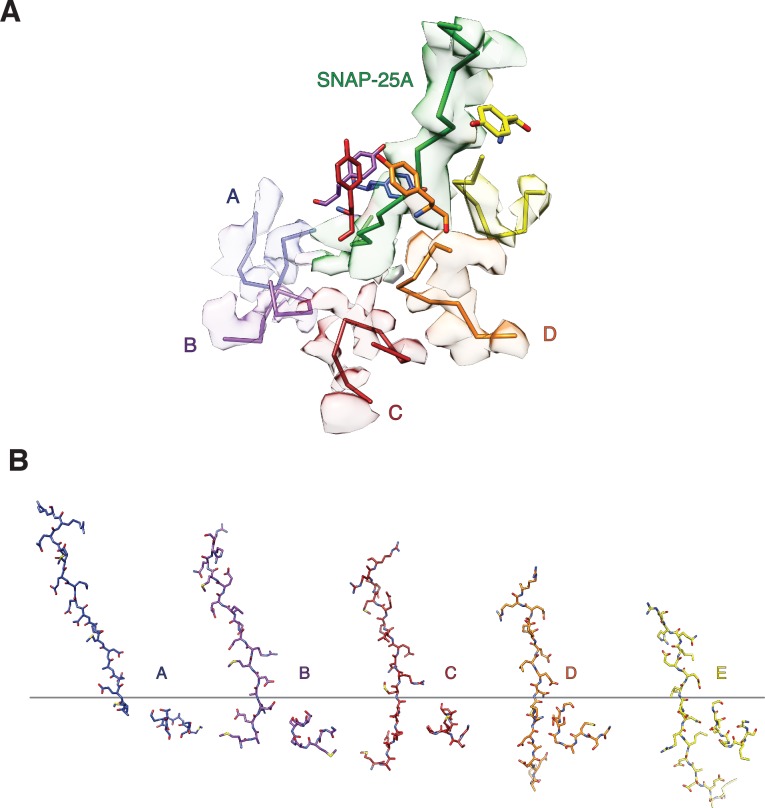
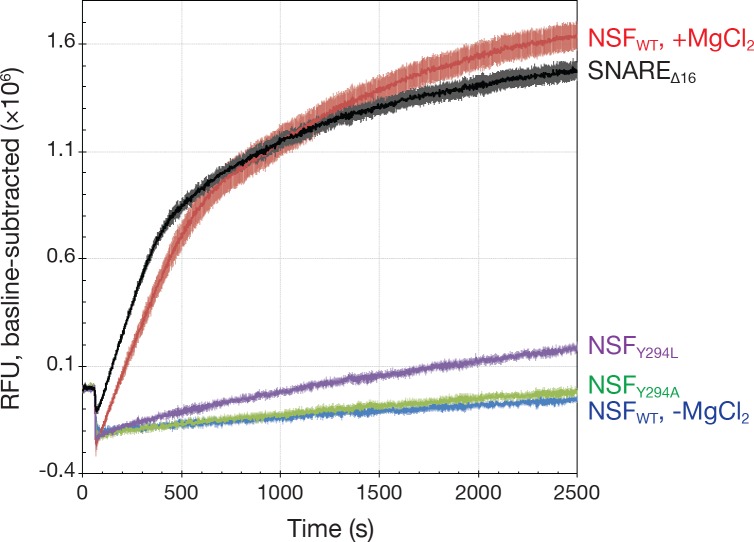
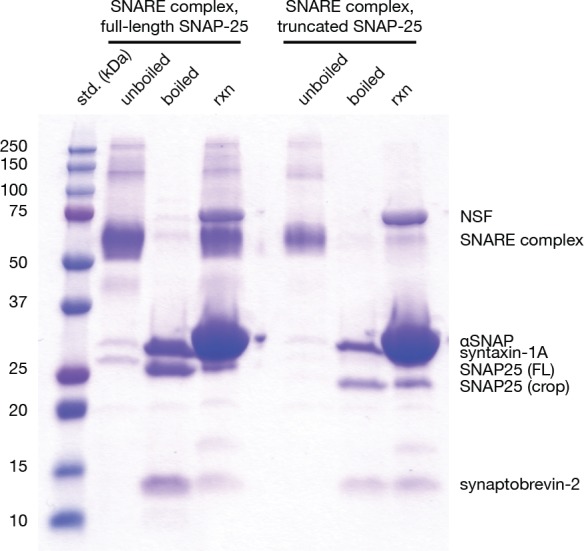
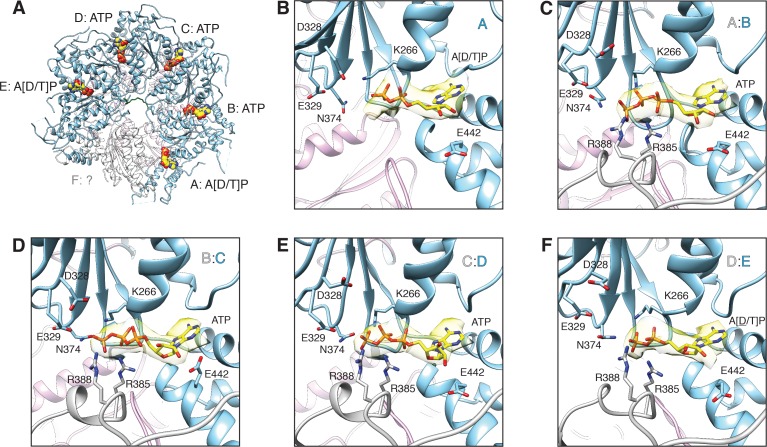
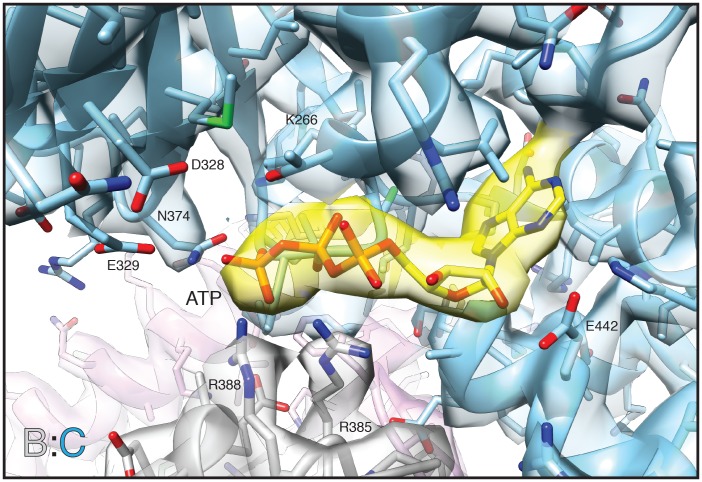
The recycling of SNARE proteins following complex formation and membrane fusion is an essential process in eukaryotic trafficking. A highly conserved AAA+ protein, NSF (N-ethylmaleimide sensitive factor) and an adaptor protein, SNAP (soluble NSF attachment protein), disassemble the SNARE complex. We report electron-cryomicroscopy structures of the complex of NSF, αSNAP, and the full-length soluble neuronal SNARE complex (composed of syntaxin-1A, synaptobrevin-2, SNAP-25A) in the presence of ATP under non-hydrolyzing conditions at ~3.9 Å resolution. These structures reveal electrostatic interactions by which two αSNAP molecules interface with a specific surface of the SNARE complex. This interaction positions the SNAREs such that the 15 N-terminal residues of SNAP-25A are loaded into the D1 ring pore of NSF via a spiral pattern of interactions between a conserved tyrosine NSF residue and SNAP-25A backbone atoms. This loading process likely precedes ATP hydrolysis. Subsequent ATP hydrolysis then drives complete disassembly.
Keywords: ATPase; SNARE complex disassembly; electron cryomicroscopy; molecular biophysics; neuroscience; neurotransmitter release; none; structural biology; synaptic vesicle recycling.
© 2018, White et al.
Conflict of interest statement
KW, MZ, UC, RP No competing interests declared, AB Reviewing editor, eLife
Figures

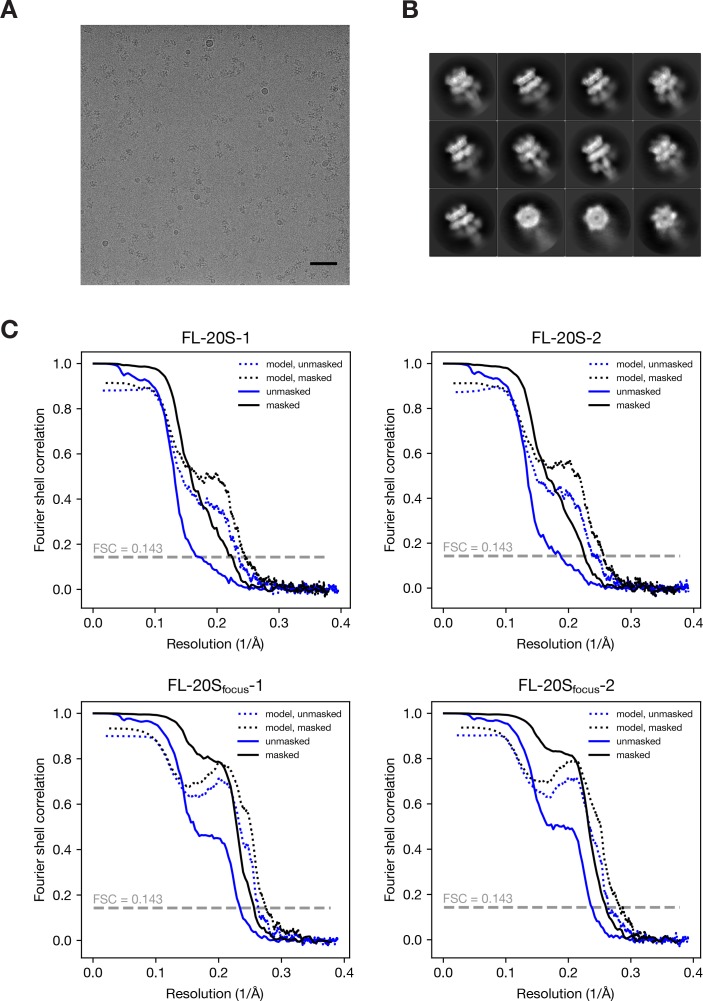
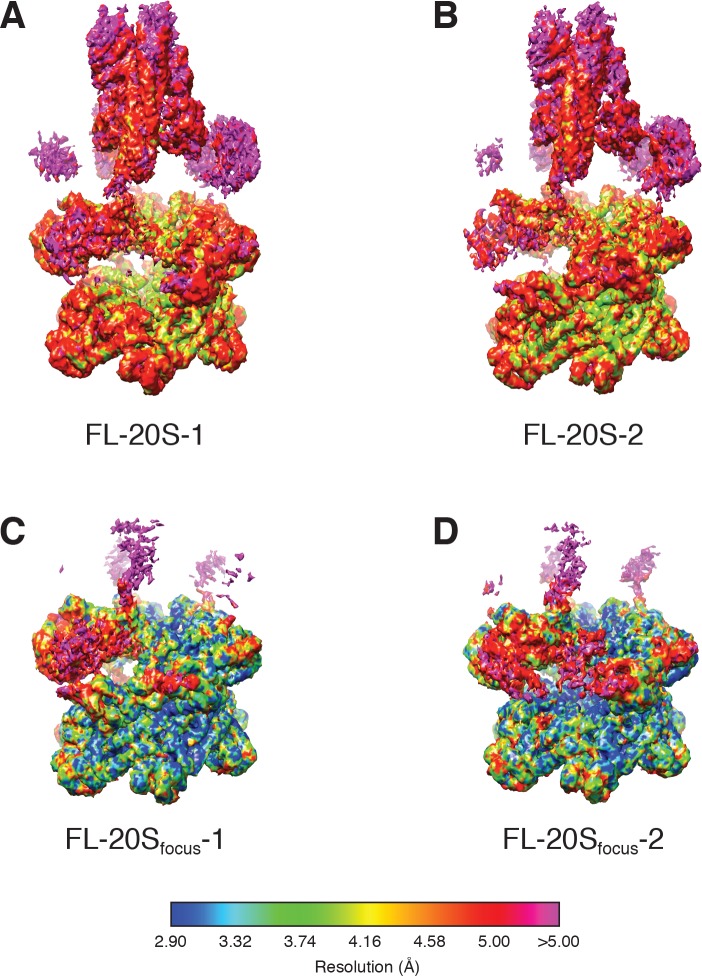
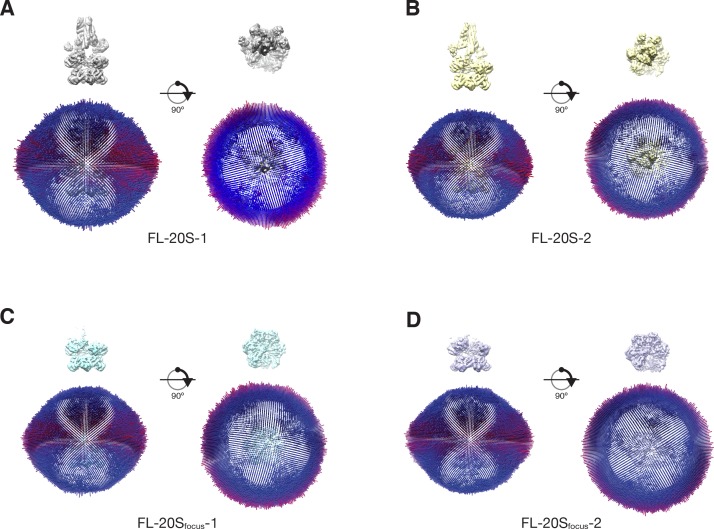
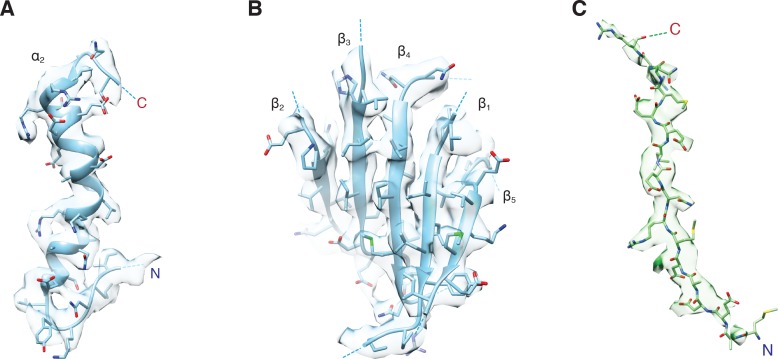
Similar articles
-
Disassembly of all SNARE complexes by N-ethylmaleimide-sensitive factor (NSF) is initiated by a conserved 1:1 interaction between α-soluble NSF attachment protein (SNAP) and SNARE complex.J Biol Chem. 2013 Aug 23;288(34):24984-91. doi: 10.1074/jbc.M113.489807. Epub 2013 Jul 8. J Biol Chem. 2013. PMID: 23836889 Free PMC article.
-
Mechanistic insights into the recycling machine of the SNARE complex.Nature. 2015 Feb 5;518(7537):61-7. doi: 10.1038/nature14148. Epub 2015 Jan 12. Nature. 2015. PMID: 25581794 Free PMC article.
-
Three αSNAP and 10 ATP molecules are used in SNARE complex disassembly by N-ethylmaleimide-sensitive factor (NSF).J Biol Chem. 2015 Jan 23;290(4):2175-88. doi: 10.1074/jbc.M114.620849. Epub 2014 Dec 9. J Biol Chem. 2015. PMID: 25492864 Free PMC article.
-
Requirements for the catalytic cycle of the N-ethylmaleimide-Sensitive Factor (NSF).Biochim Biophys Acta. 2012 Jan;1823(1):159-71. doi: 10.1016/j.bbamcr.2011.06.003. Epub 2011 Jun 13. Biochim Biophys Acta. 2012. PMID: 21689688 Free PMC article. Review.
-
Cellular functions of NSF: not just SNAPs and SNAREs.FEBS Lett. 2007 May 22;581(11):2140-9. doi: 10.1016/j.febslet.2007.03.032. Epub 2007 Mar 21. FEBS Lett. 2007. PMID: 17397838 Free PMC article. Review.
Cited by
-
Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP-dependent substrate translocation.Protein Sci. 2020 Feb;29(2):407-419. doi: 10.1002/pro.3743. Epub 2019 Oct 17. Protein Sci. 2020. PMID: 31599052 Free PMC article. Review.
-
Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex.Int J Mol Sci. 2019 Aug 1;20(15):3756. doi: 10.3390/ijms20153756. Int J Mol Sci. 2019. PMID: 31374812 Free PMC article. Review.
-
Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases.Elife. 2019 Jun 11;8:e44071. doi: 10.7554/eLife.44071. Elife. 2019. PMID: 31184588 Free PMC article.
-
Molecular Basis of VCPIP1 and P97/VCP Interaction Reveals Its Functions in Post-Mitotic Golgi Reassembly.Adv Sci (Weinh). 2024 Nov;11(41):e2403417. doi: 10.1002/advs.202403417. Epub 2024 Sep 5. Adv Sci (Weinh). 2024. PMID: 39234822 Free PMC article.
-
Structure of the human ATAD2 AAA+ histone chaperone reveals mechanism of regulation and inter-subunit communication.Commun Biol. 2023 Sep 28;6(1):993. doi: 10.1038/s42003-023-05373-1. Commun Biol. 2023. PMID: 37770645 Free PMC article.
References
-
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
