

Structural enzymology binding studies of the peptide-substrate-binding domain of human collagen prolyl 4-hydroxylase (type-II): High affinity peptides have a PxGP sequence motif
- PMID: 30168208
- PMCID: PMC6194285
- DOI: 10.1002/pro.3450
Structural enzymology binding studies of the peptide-substrate-binding domain of human collagen prolyl 4-hydroxylase (type-II): High affinity peptides have a PxGP sequence motif
Abstract
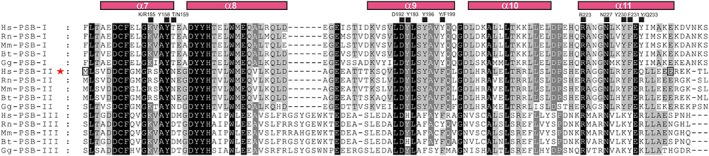
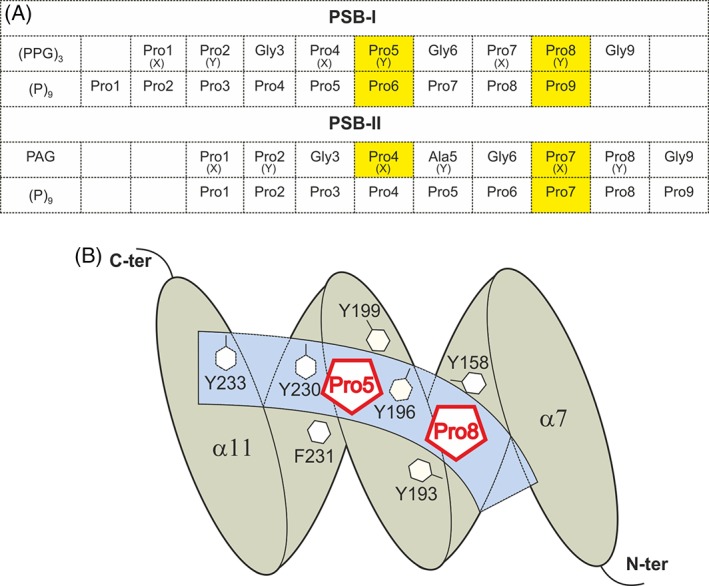
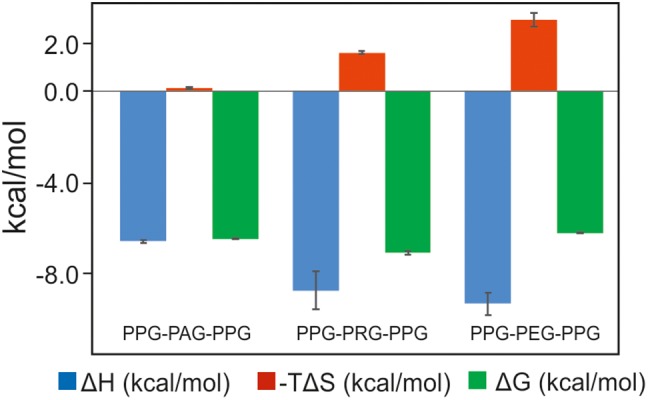
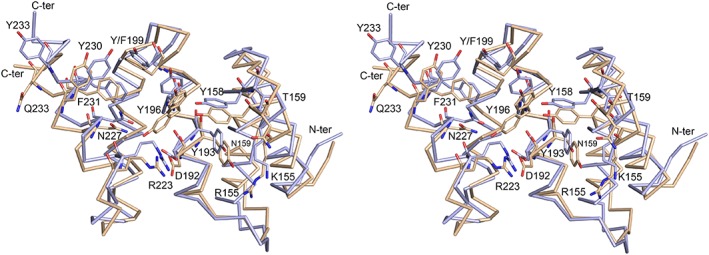
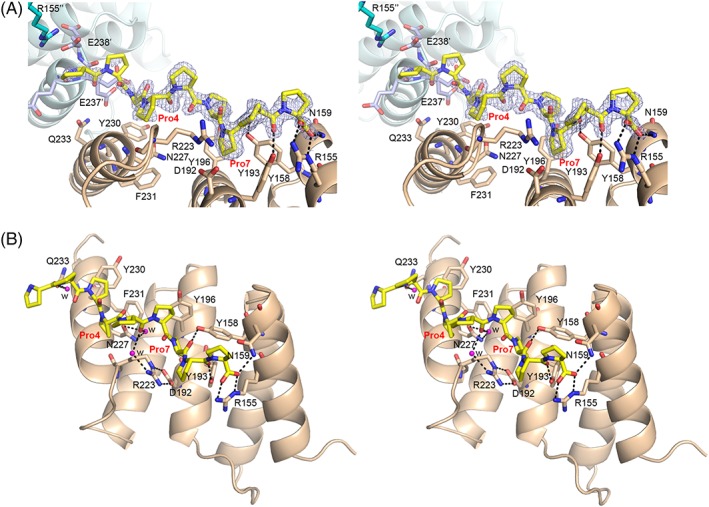
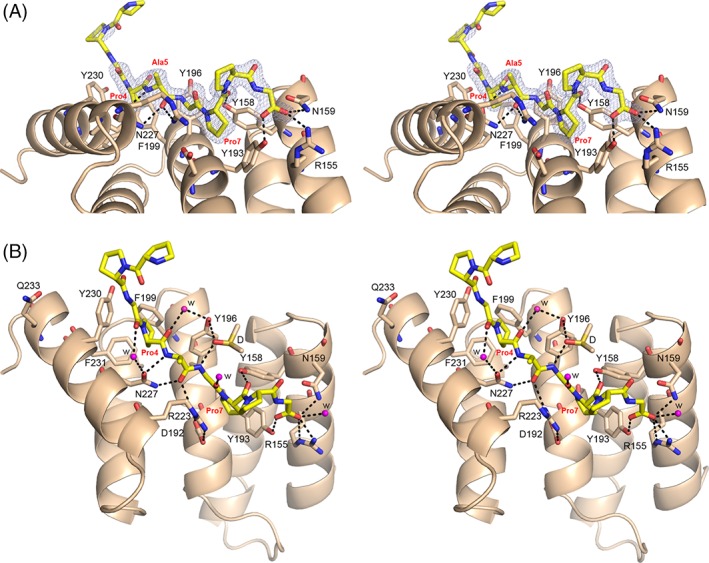
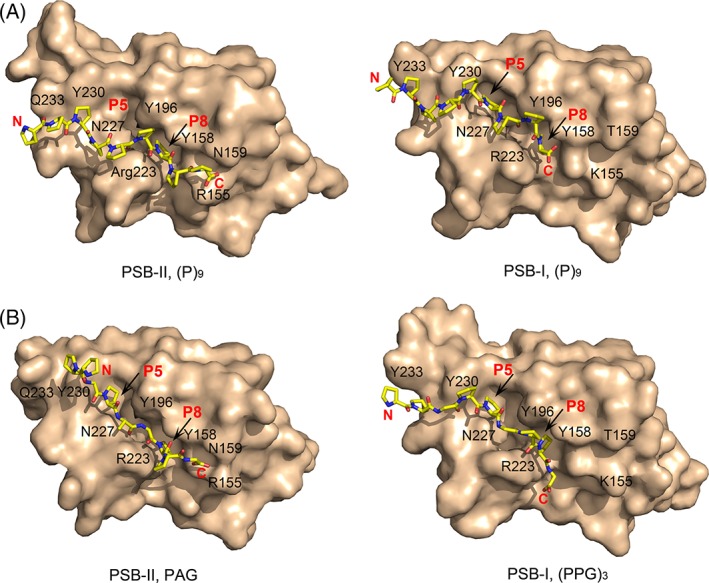
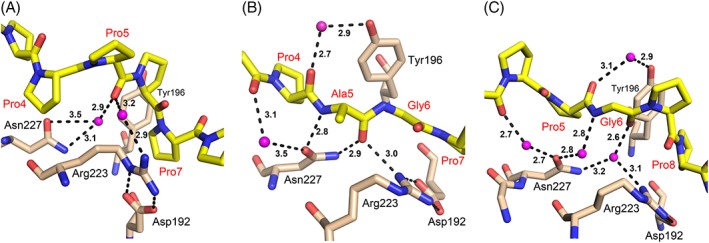
The peptide-substrate-binding (PSB) domain of collagen prolyl 4-hydroxylase (C-P4H, an α2 β2 tetramer) binds proline-rich procollagen peptides. This helical domain (the middle domain of the α subunit) has an important role concerning the substrate binding properties of C-P4H, although it is not known how the PSB domain influences the hydroxylation properties of the catalytic domain (the C-terminal domain of the α subunit). The crystal structures of the PSB domain of the human C-P4H isoform II (PSB-II) complexed with and without various short proline-rich peptides are described. The comparison with the previously determined PSB-I peptide complex structures shows that the C-P4H-I substrate peptide (PPG)3 , has at most very weak affinity for PSB-II, although it binds with high affinity to PSB-I. The replacement of the middle PPG triplet of (PPG)3 to the nonhydroxylatable PAG, PRG, or PEG triplet, increases greatly the affinity of PSB-II for these peptides, leading to a deeper mode of binding, as compared to the previously determined PSB-I peptide complexes. In these PSB-II complexes, the two peptidyl prolines of its central P(A/R/E)GP region bind in the Pro5 and Pro8 binding pockets of the PSB peptide-binding groove, and direct hydrogen bonds are formed between the peptide and the side chains of the highly conserved residues Tyr158, Arg223, and Asn227, replacing water mediated interactions in the corresponding PSB-I complex. These results suggest that PxGP (where x is not a proline) is the common motif of proline-rich peptide sequences that bind with high affinity to PSB-II.
Keywords: X-ray crystallography; calorimetry; collagen; extracellular matrix protein; proline-rich peptide; prolyl 4-hydroxylase; protein-peptide interactions.
© 2018 The Protein Society.
Figures
Similar articles
-
Assembly of the elongated collagen prolyl 4-hydroxylase α2β2 heterotetramer around a central α2 dimer.Biochem J. 2017 Feb 20;474(5):751-769. doi: 10.1042/BCJ20161000. Biochem J. 2017. PMID: 28093469
-
Crystal structure of the collagen prolyl 4-hydroxylase (C-P4H) catalytic domain complexed with PDI: Toward a model of the C-P4H α2β2 tetramer.J Biol Chem. 2022 Dec;298(12):102614. doi: 10.1016/j.jbc.2022.102614. Epub 2022 Oct 18. J Biol Chem. 2022. PMID: 36265586 Free PMC article.
-
The structural motifs for substrate binding and dimerization of the α subunit of collagen prolyl 4-hydroxylase.Structure. 2013 Dec 3;21(12):2107-18. doi: 10.1016/j.str.2013.09.005. Epub 2013 Oct 24. Structure. 2013. PMID: 24207127
-
Protein hydroxylation: prolyl 4-hydroxylase, an enzyme with four cosubstrates and a multifunctional subunit.FASEB J. 1989 Mar;3(5):1609-17. FASEB J. 1989. PMID: 2537773 Review.
-
Collagen prolyl 4-hydroxylases modify tumor progression.Acta Biochim Biophys Sin (Shanghai). 2021 Jul 5;53(7):805-814. doi: 10.1093/abbs/gmab065. Acta Biochim Biophys Sin (Shanghai). 2021. PMID: 34009234 Review.
Cited by
-
IceBear: an intuitive and versatile web application for research-data tracking from crystallization experiment to PDB deposition.Acta Crystallogr D Struct Biol. 2021 Feb 1;77(Pt 2):151-163. doi: 10.1107/S2059798320015223. Epub 2021 Jan 26. Acta Crystallogr D Struct Biol. 2021. PMID: 33559605 Free PMC article.
-
Role of prolyl hydroxylation in the molecular interactions of collagens.Essays Biochem. 2019 Sep 13;63(3):325-335. doi: 10.1042/EBC20180053. Print 2019 Sep 13. Essays Biochem. 2019. PMID: 31350381 Free PMC article. Review.
-
P4HA1 as an unfavorable prognostic marker promotes cell migration and invasion of glioblastoma via inducing EMT process under hypoxia microenvironment.Am J Cancer Res. 2021 Feb 1;11(2):590-617. eCollection 2021. Am J Cancer Res. 2021. PMID: 33575089 Free PMC article.
-
Computed Protein-Protein Enthalpy Signatures as a Tool for Identifying Conformation Sampling Problems.J Chem Inf Model. 2023 Oct 9;63(19):6095-6108. doi: 10.1021/acs.jcim.3c01041. Epub 2023 Sep 27. J Chem Inf Model. 2023. PMID: 37759363 Free PMC article.
-
Glycosylation Modulates the Structure and Functions of Collagen: A Review.Molecules. 2024 Mar 22;29(7):1417. doi: 10.3390/molecules29071417. Molecules. 2024. PMID: 38611696 Free PMC article. Review.
References
-
- Ball LJ, Kühne R, Schneider‐Mergener J, Oschkinat H (2005) Recognition of proline‐rich motifs by protein‐protein‐interaction domains. Angew Chem Int Ed Engl 44:2852–2869. - PubMed
-
- Bhattacharyya R, Chakrabarti P (2003) Stereospecific interactions of proline residues in protein structures and complexes. J Mol Biol 331:925–940. - PubMed
-
- Opitz R, Müller M, Reuter C, Barone M, Soicke A, Roske Y, Piotukh K, Huy P, Beerbaum M, Wiesner B, Beyermann M, Schmieder P, Freund C, Volkmer R, Oschkinat H, Schmalz HG, Kühne R (2015) A modular toolkit to inhibit proline‐rich motif‐mediated protein‐protein interactions. Proc Natl Acad Sci USA 112:5011–5016. - PMC - PubMed
-
- Cubellis MV, Caillez F, Blundell TL, Lovel SC (2005) Properties of polyproline II, a secondary structure element implicated in protein‐protein interactions. Proteins 58:880–892. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
