

The Epstein Barr virus circRNAome
- PMID: 30080890
- PMCID: PMC6095625
- DOI: 10.1371/journal.ppat.1007206
The Epstein Barr virus circRNAome
Abstract
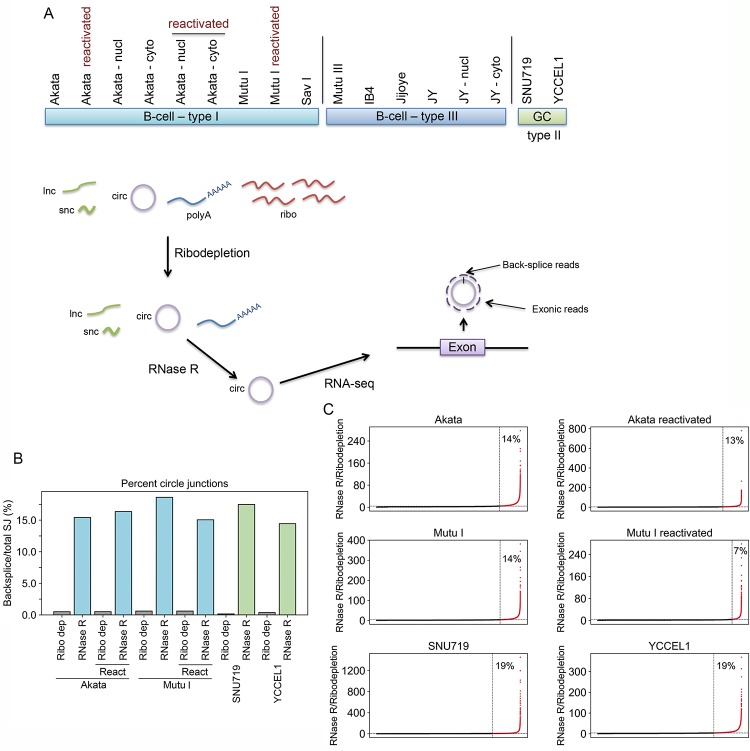
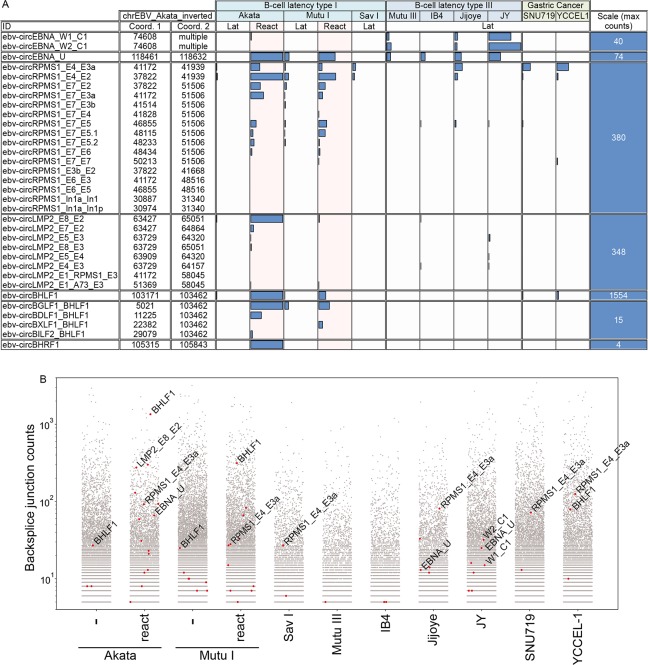
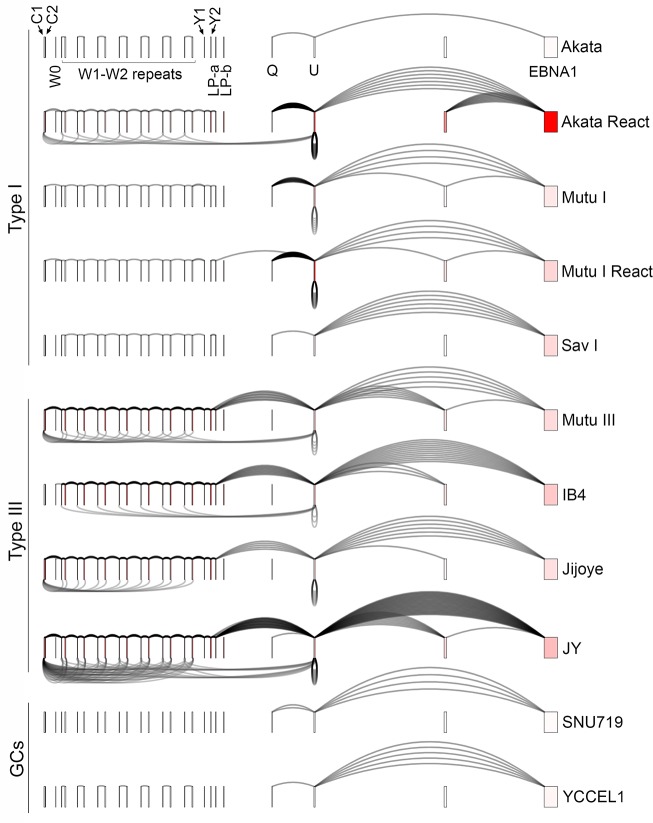
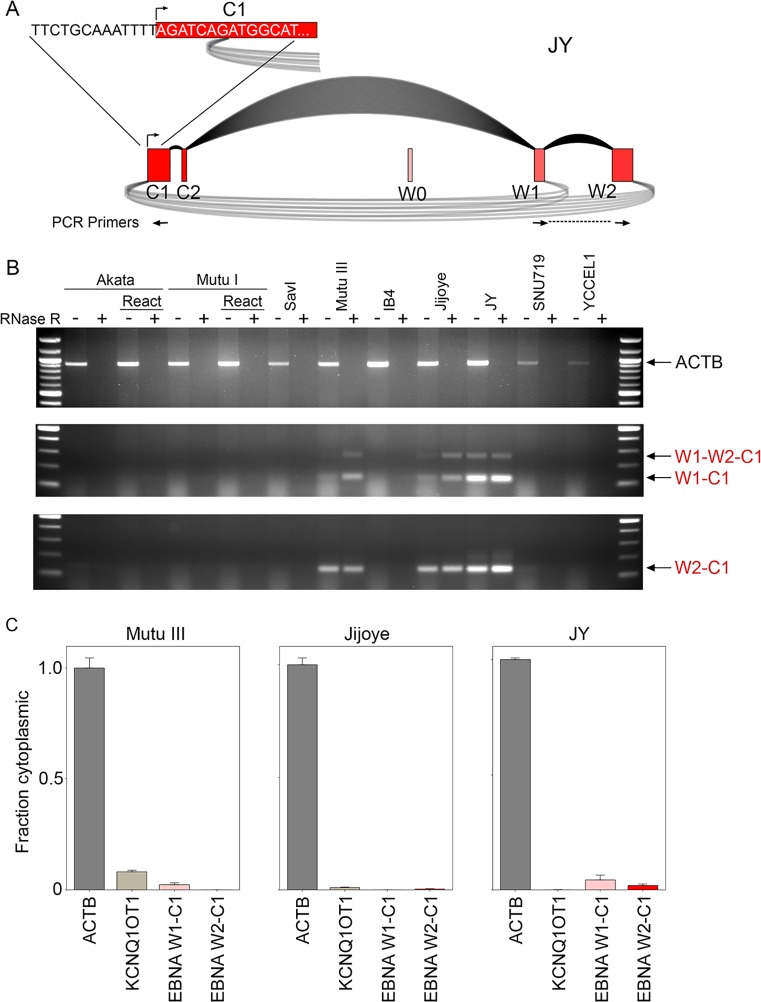
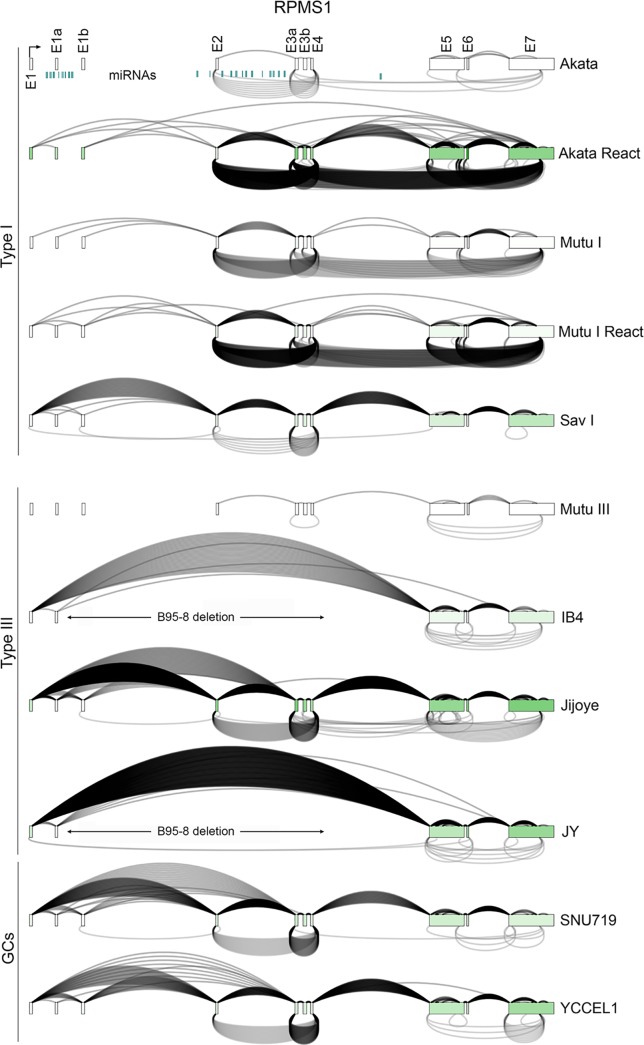
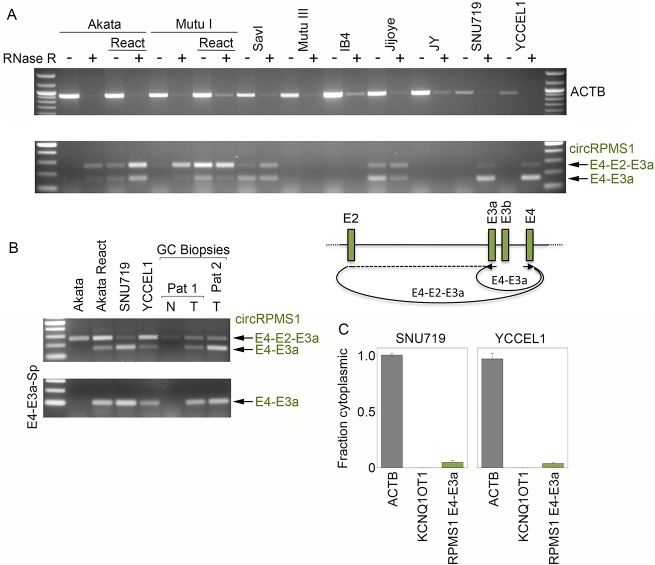
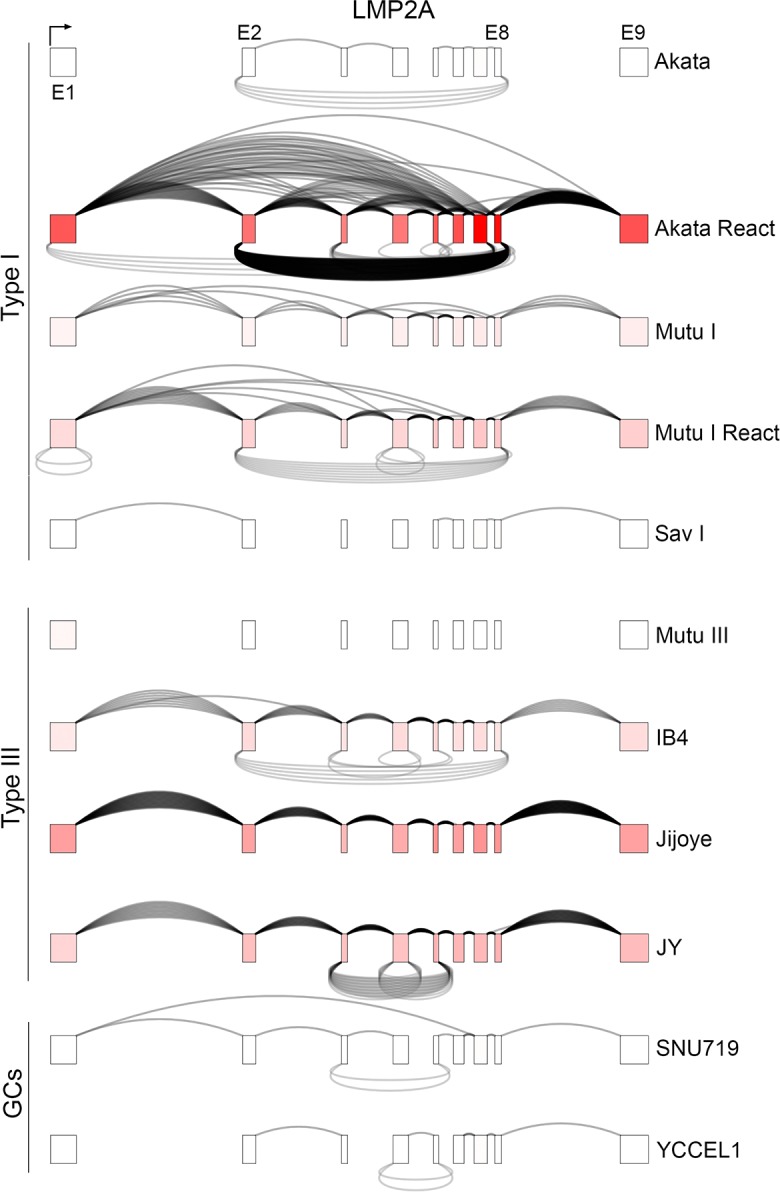
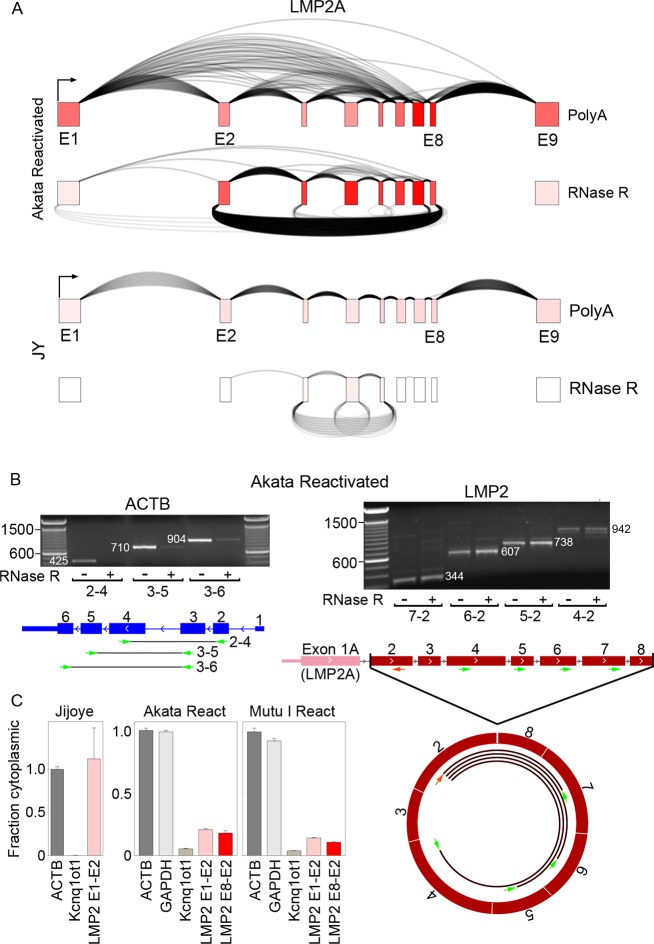
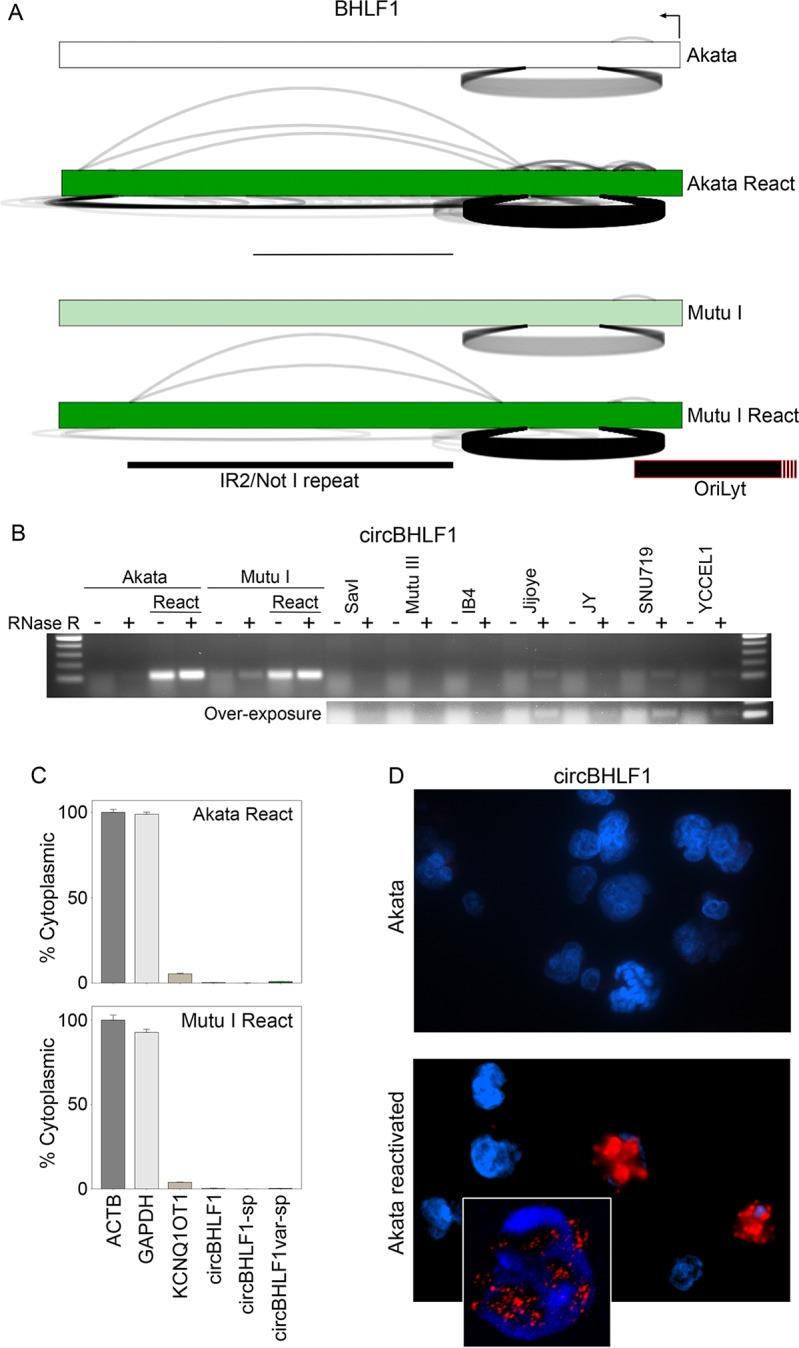
Our appreciation for the extent of Epstein Barr virus (EBV) transcriptome complexity continues to grow through findings of EBV encoded microRNAs, new long non-coding RNAs as well as the more recent discovery of over a hundred new polyadenylated lytic transcripts. Here we report an additional layer to the EBV transcriptome through the identification of a repertoire of latent and lytic viral circular RNAs. Utilizing RNase R-sequencing with cell models representing latency types I, II, and III, we identified EBV encoded circular RNAs expressed from the latency Cp promoter involving backsplicing from the W1 and W2 exons to the C1 exon, from the EBNA BamHI U fragment exon, and from the latency long non-coding RPMS1 locus. In addition, we identified circular RNAs expressed during reactivation including backsplicing from exon 8 to exon 2 of the LMP2 gene and a highly expressed circular RNA derived from intra-exonic backsplicing within the BHLF1 gene. While expression of most of these circular RNAs was found to depend on the EBV transcriptional program utilized and the transcription levels of the associated loci, expression of LMP2 exon 8 to exon 2 circular RNA was found to be cell model specific. Altogether we identified over 30 unique EBV circRNAs candidates and we validated and determined the structural features, expression profiles and nuclear/cytoplasmic distributions of several predominant and notable viral circRNAs. Further, we show that two of the EBV circular RNAs derived from the RPMS1 locus are detected in EBV positive clinical stomach cancer specimens. This study increases the known EBV latency and lytic transcriptome repertoires to include viral circular RNAs and it provides an essential foundation and resource for investigations into the functions and roles of this new class of EBV transcripts in EBV biology and diseases.
Conflict of interest statement
The authors, TL and MS, are currently under the employment of ReproCELL Incorporated. ReproCELL Incorporated provided funds, resources and equipment to conduct some of the studies and provided salary and benefits for TL and MS. This does not alter our adherence to all PLoS Pathogens policies on sharing data and materials.
Figures
Similar articles
-
Comparative Analysis of Gammaherpesvirus Circular RNA Repertoires: Conserved and Unique Viral Circular RNAs.J Virol. 2019 Mar 5;93(6):e01952-18. doi: 10.1128/JVI.01952-18. Print 2019 Mar 15. J Virol. 2019. PMID: 30567979 Free PMC article.
-
Circular DNA tumor viruses make circular RNAs.Proc Natl Acad Sci U S A. 2018 Sep 11;115(37):E8737-E8745. doi: 10.1073/pnas.1811728115. Epub 2018 Aug 27. Proc Natl Acad Sci U S A. 2018. PMID: 30150410 Free PMC article.
-
Identification and characterization of a novel Epstein-Barr Virus-encoded circular RNA from LMP-2 Gene.Sci Rep. 2021 Jul 13;11(1):14392. doi: 10.1038/s41598-021-93781-w. Sci Rep. 2021. PMID: 34257379 Free PMC article.
-
EBV Noncoding RNAs.Curr Top Microbiol Immunol. 2015;391:181-217. doi: 10.1007/978-3-319-22834-1_6. Curr Top Microbiol Immunol. 2015. PMID: 26428375 Free PMC article. Review.
-
Regulation and dysregulation of Epstein-Barr virus latency: implications for the development of autoimmune diseases.Autoimmunity. 2008 May;41(4):298-328. doi: 10.1080/08916930802024772. Autoimmunity. 2008. PMID: 18432410 Review.
Cited by
-
A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer.Oncogene. 2019 Jun;38(25):4977-4989. doi: 10.1038/s41388-019-0768-8. Epub 2019 Feb 28. Oncogene. 2019. PMID: 30820039 Free PMC article.
-
Functions of Circular RNA in Human Diseases and Illnesses.Noncoding RNA. 2023 Jul 4;9(4):38. doi: 10.3390/ncrna9040038. Noncoding RNA. 2023. PMID: 37489458 Free PMC article. Review.
-
Role of Virus-Induced Host Cell Epigenetic Changes in Cancer.Int J Mol Sci. 2021 Aug 3;22(15):8346. doi: 10.3390/ijms22158346. Int J Mol Sci. 2021. PMID: 34361112 Free PMC article. Review.
-
Epstein-Barr Virus-Encoded Circular RNA CircBART2.2 Promotes Immune Escape of Nasopharyngeal Carcinoma by Regulating PD-L1.Cancer Res. 2021 Oct 1;81(19):5074-5088. doi: 10.1158/0008-5472.CAN-20-4321. Epub 2021 Jul 28. Cancer Res. 2021. PMID: 34321242 Free PMC article.
-
Identifying and characterizing virus-encoded circular RNAs.Methods. 2021 Dec;196:129-137. doi: 10.1016/j.ymeth.2021.03.004. Epub 2021 Mar 11. Methods. 2021. PMID: 33713796 Free PMC article. Review.
References
-
- Longnecker R, Kieff E, Cohen J. Epstein-Barr virus. 6th ed. DM K, PM H, editors. Philadelphia, PA2013.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
