

The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation
- PMID: 30020827
- PMCID: PMC6230687
- DOI: 10.1152/ajpcell.00098.2018
The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation
Abstract
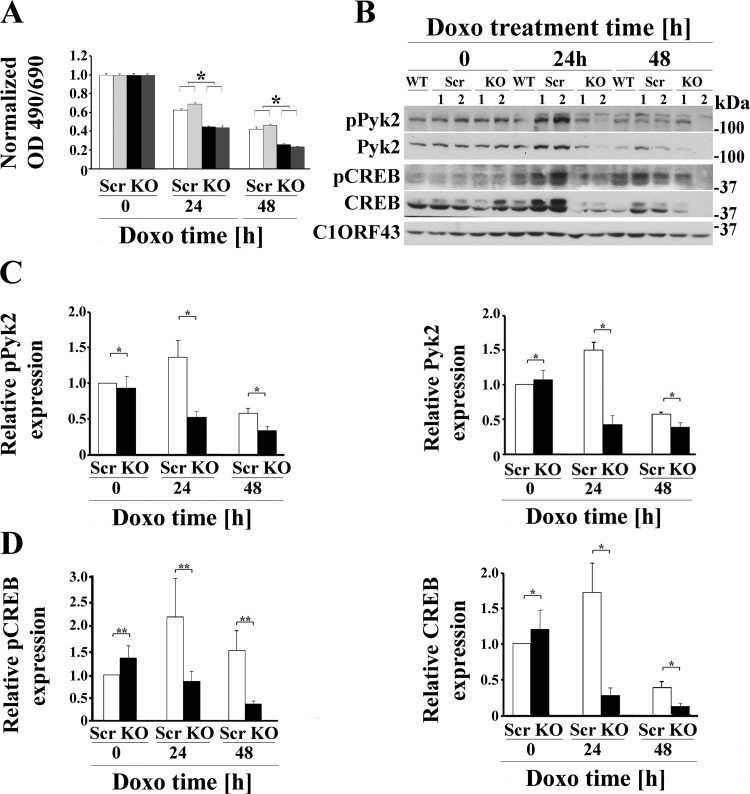
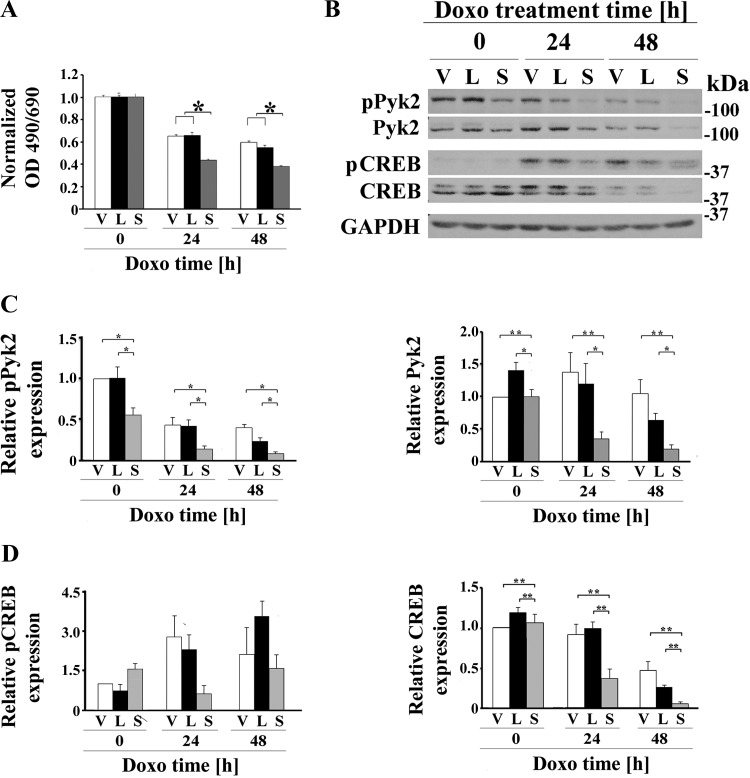
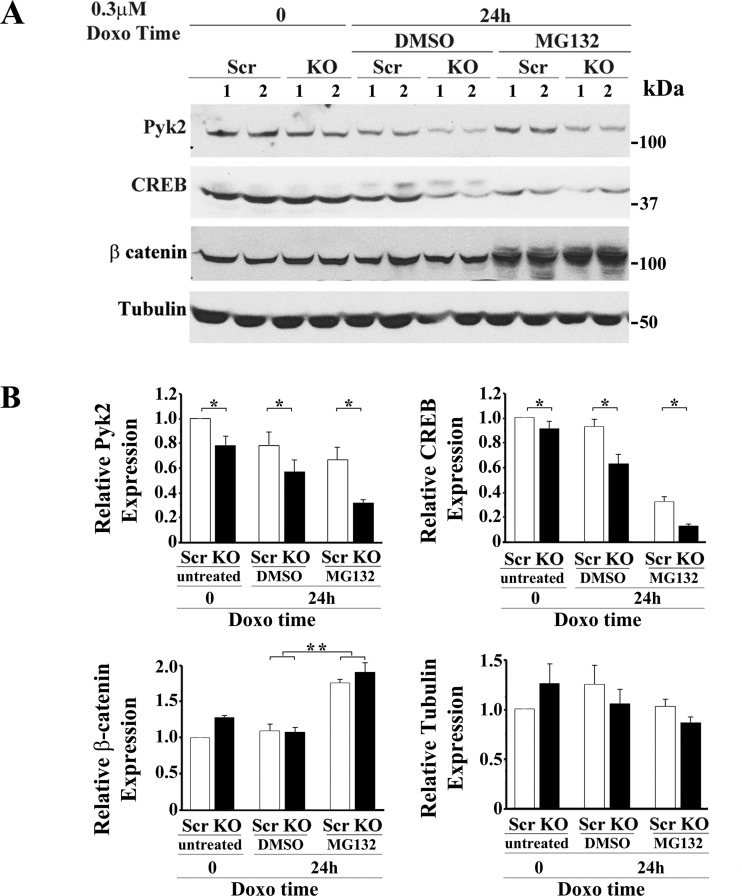
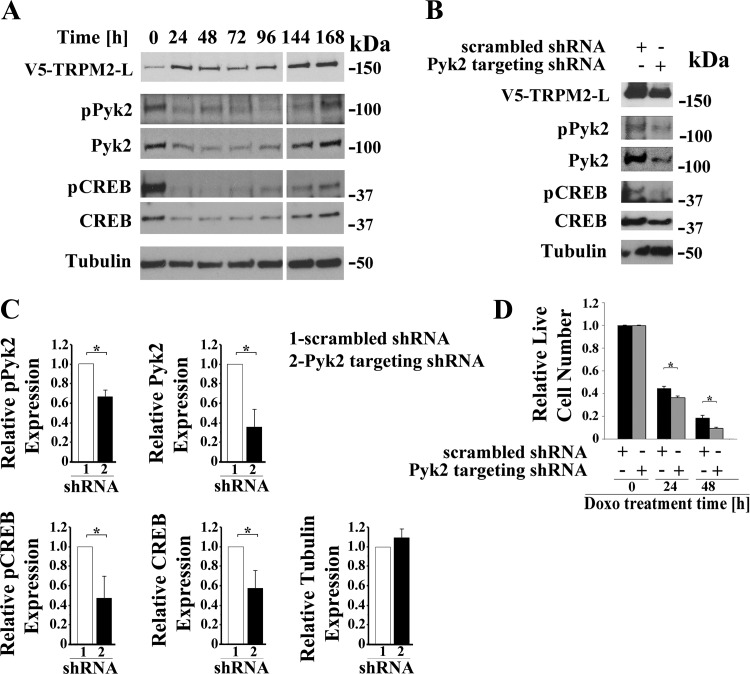
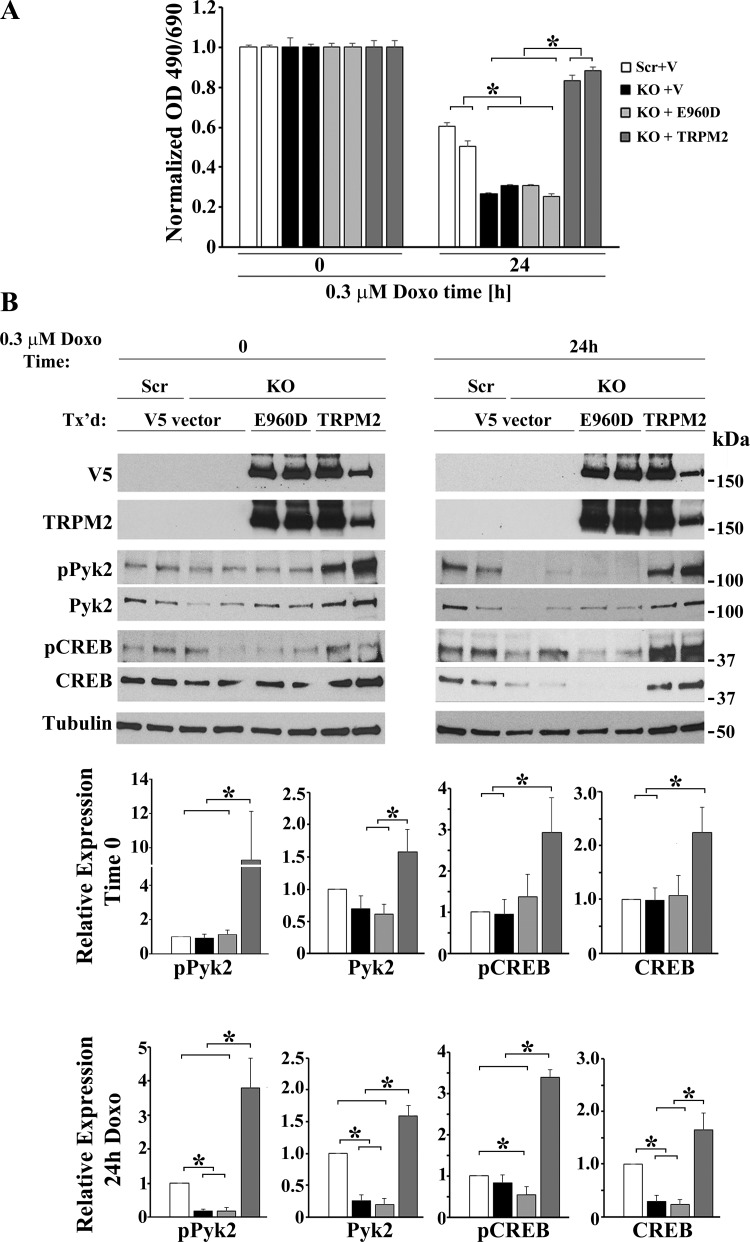
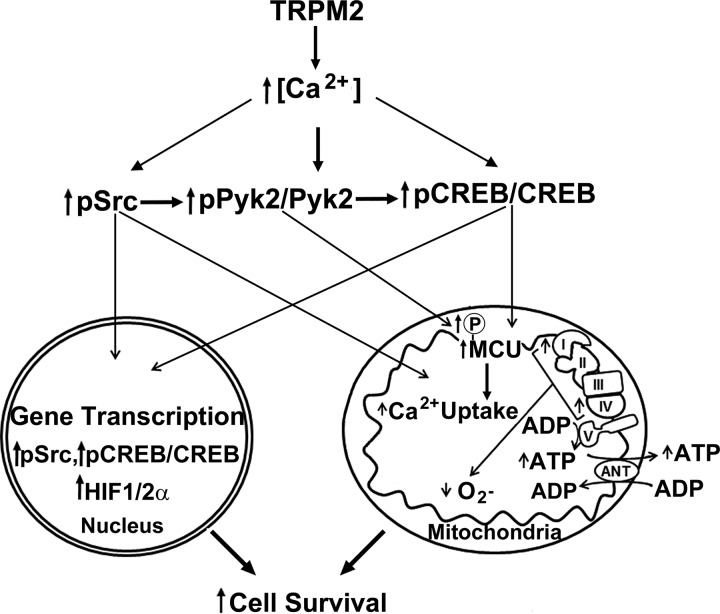
Transient receptor potential melastatin channel subfamily member 2 (TRPM2) has an essential function in cell survival and is highly expressed in many cancers. Inhibition of TRPM2 in neuroblastoma by depletion with CRISPR technology or expression of dominant negative TRPM2-S has been shown to significantly reduce cell viability. Here, the role of proline-rich tyrosine kinase 2 (Pyk2) in TRPM2 modulation of neuroblastoma viability was explored. In TRPM2-depleted cells, phosphorylation and expression of Pyk2 and cAMP-responsive element-binding protein (CREB), a downstream target, were significantly reduced after application of the chemotherapeutic agent doxorubicin. Overexpression of wild-type Pyk2 rescued cell viability. Reduction of Pyk2 expression with shRNA decreased cell viability and CREB phosphorylation and expression, demonstrating Pyk2 modulates CREB activation. TRPM2 depletion impaired phosphorylation of Src, an activator of Pyk2, and this may be a mechanism to reduce Pyk2 phosphorylation. TRPM2 inhibition was previously demonstrated to decrease mitochondrial function. Here, CREB, Pyk2, and phosphorylated Src were reduced in mitochondria of TRPM2-depleted cells, consistent with their role in modulating expression and activation of mitochondrial proteins. Phosphorylated Src and phosphorylated and total CREB were reduced in TRPM2-depleted nuclei. Expression and function of mitochondrial calcium uniporter (MCU), a target of phosphorylated Pyk2 and CREB, were significantly reduced. Wild-type TRPM2 but not Ca2+-impermeable mutant E960D reconstituted phosphorylation and expression of Pyk2 and CREB in TRPM2-depleted cells exposed to doxorubicin. Results demonstrate that TRPM2 expression protects the viability of neuroblastoma through Src, Pyk2, CREB, and MCU activation, which play key roles in maintaining mitochondrial function and cellular bioenergetics.
Keywords: CREB; MCU; Pyk2; ROS; Src; TRPM2; mitochondria; neuroblastoma.
Figures

Similar articles
-
Depletion of the Human Ion Channel TRPM2 in Neuroblastoma Demonstrates Its Key Role in Cell Survival through Modulation of Mitochondrial Reactive Oxygen Species and Bioenergetics.J Biol Chem. 2016 Nov 18;291(47):24449-24464. doi: 10.1074/jbc.M116.747147. Epub 2016 Sep 30. J Biol Chem. 2016. PMID: 27694440 Free PMC article.
-
TRPM2 in Cancer.Cell Calcium. 2019 Jun;80:8-17. doi: 10.1016/j.ceca.2019.03.002. Epub 2019 Mar 6. Cell Calcium. 2019. PMID: 30925291 Free PMC article. Review.
-
Transient receptor potential ion channel TRPM2 promotes AML proliferation and survival through modulation of mitochondrial function, ROS, and autophagy.Cell Death Dis. 2020 Apr 20;11(4):247. doi: 10.1038/s41419-020-2454-8. Cell Death Dis. 2020. PMID: 32312983 Free PMC article.
-
A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2α.J Biol Chem. 2014 Dec 26;289(52):36284-302. doi: 10.1074/jbc.M114.620922. Epub 2014 Nov 12. J Biol Chem. 2014. PMID: 25391657 Free PMC article.
-
Transient Receptor Potential-Melastatin Channel Family Member 2: Friend or Foe.Trans Am Clin Climatol Assoc. 2017;128:308-329. Trans Am Clin Climatol Assoc. 2017. PMID: 28790515 Free PMC article. Review.
Cited by
-
Pyk2/MCU Pathway as a New Target for Reversing Atherosclerosis.Front Cell Dev Biol. 2021 May 6;9:651579. doi: 10.3389/fcell.2021.651579. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34026753 Free PMC article.
-
TRPM2 Channels: A Potential Therapeutic Target in Melanoma?Int J Mol Sci. 2023 Jun 21;24(13):10437. doi: 10.3390/ijms241310437. Int J Mol Sci. 2023. PMID: 37445615 Free PMC article. Review.
-
Mitochondrial translation failure represses cholesterol gene expression via Pyk2-Gsk3β-Srebp2 axis.Life Sci Alliance. 2024 May 8;7(7):e202302423. doi: 10.26508/lsa.202302423. Print 2024 Jul. Life Sci Alliance. 2024. PMID: 38719751 Free PMC article.
-
Advances in the role of ion channels in leukemia.Heliyon. 2024 Jun 22;10(12):e33452. doi: 10.1016/j.heliyon.2024.e33452. eCollection 2024 Jun 30. Heliyon. 2024. PMID: 39027429 Free PMC article. Review.
-
TRPM2 ion channel promotes gastric cancer migration, invasion and tumor growth through the AKT signaling pathway.Sci Rep. 2019 Mar 12;9(1):4182. doi: 10.1038/s41598-019-40330-1. Sci Rep. 2019. PMID: 30862883 Free PMC article.
References
-
- Almasi S, Kennedy BE, El-Aghil M, Sterea AM, Gujar S, Partida-Sánchez S, El Hiani Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem 293: 3637–3650, 2018. doi:10.1074/jbc.M117.817635. - DOI - PMC - PubMed
-
- Banno Y, Nemoto S, Murakami M, Kimura M, Ueno Y, Ohguchi K, Hara A, Okano Y, Kitade Y, Onozuka M, Murate T, Nozawa Y. Depolarization-induced differentiation of PC12 cells is mediated by phospholipase D2 through the transcription factor CREB pathway. J Neurochem 104: 1372–1386, 2008. doi:10.1111/j.1471-4159.2007.05085.x. - DOI - PubMed
-
- Bao L, Chen SJ, Conrad K, Keefer K, Abraham T, Lee JP, Wang J, Zhang XQ, Hirschler-Laszkiewicz I, Wang HG, Dovat S, Gans B, Madesh M, Cheung JY, Miller BA. depletion of the human ion channel TRPM2 in neuroblastoma demonstrates its key role in cell survival through modulation of mitochondrial reactive oxygen species and bioenergetics. J Biol Chem 291: 24449–24464, 2016. doi:10.1074/jbc.M116.747147. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous