

Virus DNA Replication and the Host DNA Damage Response
- PMID: 29996066
- PMCID: PMC6462412
- DOI: 10.1146/annurev-virology-092917-043534
Virus DNA Replication and the Host DNA Damage Response
Abstract
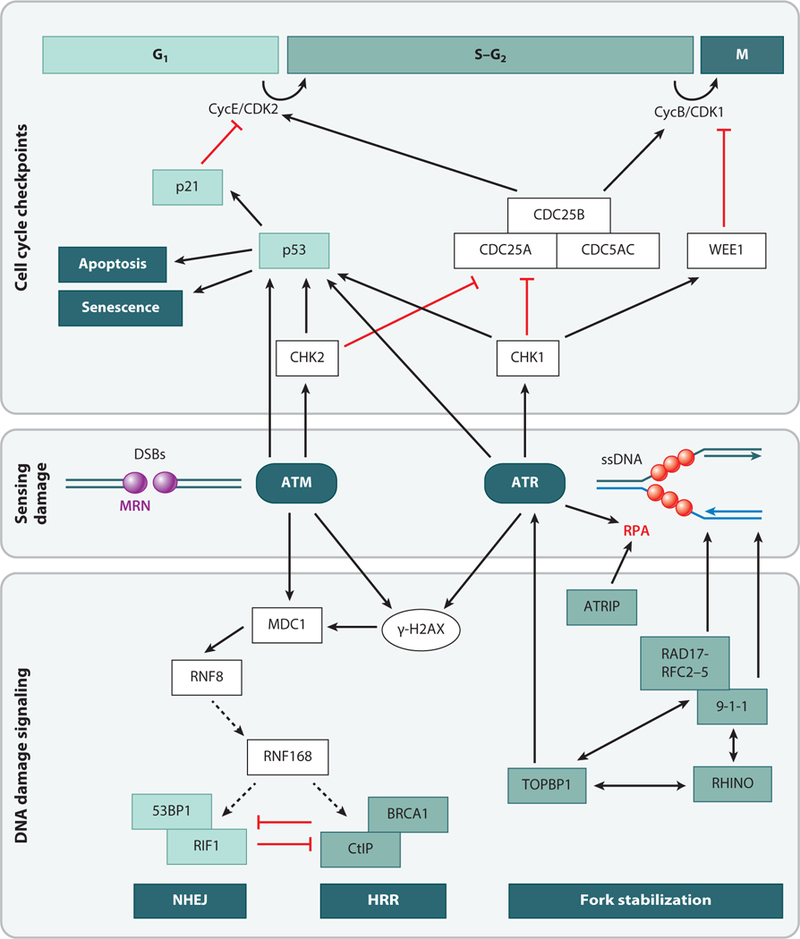
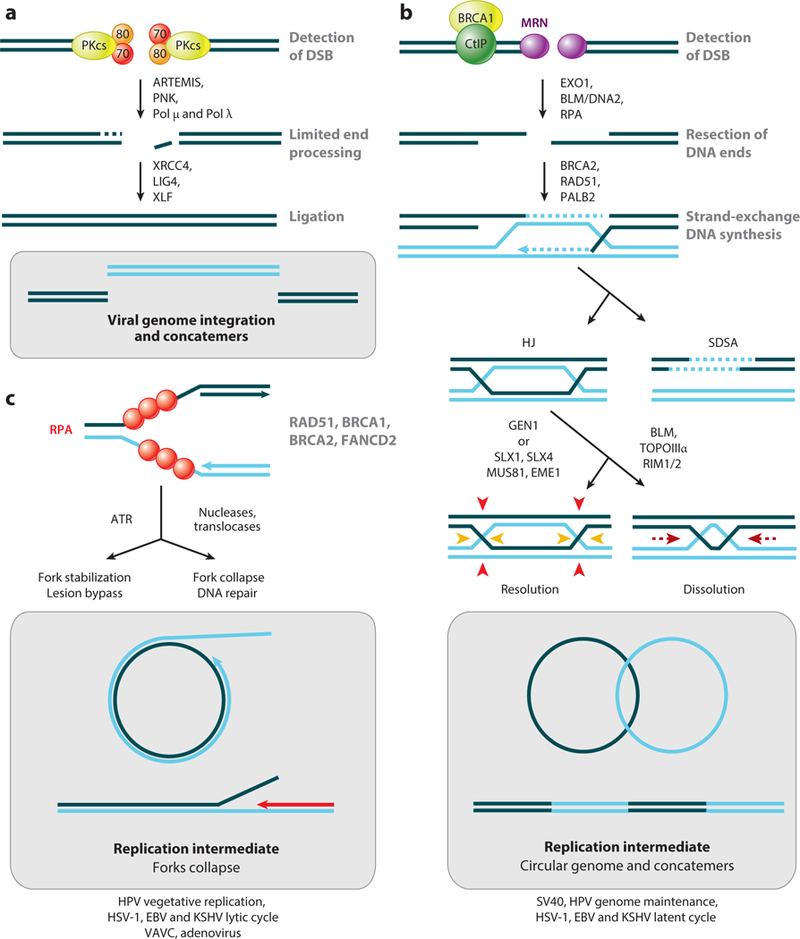
Viral DNA genomes have limited coding capacity and therefore harness cellular factors to facilitate replication of their genomes and generate progeny virions. Studies of viruses and how they interact with cellular processes have historically provided seminal insights into basic biology and disease mechanisms. The replicative life cycles of many DNA viruses have been shown to engage components of the host DNA damage and repair machinery. Viruses have evolved numerous strategies to navigate the cellular DNA damage response. By hijacking and manipulating cellular replication and repair processes, DNA viruses can selectively harness or abrogate distinct components of the cellular machinery to complete their life cycles. Here, we highlight consequences for viral replication and host genome integrity during the dynamic interactions between virus and host.
Keywords: DNA damage response; chromatin state; viral genome; virus replication; virus replication compartments.
Figures
Similar articles
-
DNA viruses and the cellular DNA-damage response.J Gen Virol. 2012 Oct;93(Pt 10):2076-2097. doi: 10.1099/vir.0.044412-0. Epub 2012 Aug 1. J Gen Virol. 2012. PMID: 22855786 Review.
-
Mechanisms of Host IFI16, PML, and Daxx Protein Restriction of Herpes Simplex Virus 1 Replication.J Virol. 2018 Apr 27;92(10):e00057-18. doi: 10.1128/JVI.00057-18. Print 2018 May 15. J Virol. 2018. PMID: 29491153 Free PMC article.
-
Playing with fire: consequences of human papillomavirus DNA replication adjacent to genetically unstable regions of host chromatin.Curr Opin Virol. 2017 Oct;26:63-68. doi: 10.1016/j.coviro.2017.07.015. Epub 2017 Aug 3. Curr Opin Virol. 2017. PMID: 28779692 Review.
-
Interactions of viruses with the cellular DNA repair machinery.DNA Repair (Amst). 2004 Aug-Sep;3(8-9):1165-73. doi: 10.1016/j.dnarep.2004.03.018. DNA Repair (Amst). 2004. PMID: 15279805 Review.
-
Activation of the DNA Damage Response by RNA Viruses.Biomolecules. 2016 Jan 6;6(1):2. doi: 10.3390/biom6010002. Biomolecules. 2016. PMID: 26751489 Free PMC article. Review.
Cited by
-
Early Nuclear Events after Herpesviral Infection.J Clin Med. 2019 Sep 7;8(9):1408. doi: 10.3390/jcm8091408. J Clin Med. 2019. PMID: 31500286 Free PMC article. Review.
-
The consequences of viral infection on host DNA damage response: a focus on SARS-CoVs.J Genet Eng Biotechnol. 2022 Jul 13;20(1):104. doi: 10.1186/s43141-022-00388-3. J Genet Eng Biotechnol. 2022. PMID: 35829826 Free PMC article. Review.
-
Deleterious and ethnic-related BRCA1/2 mutations in tissue and blood of Egyptian colorectal cancer patients and its correlation with human papillomavirus.Clin Exp Med. 2023 Dec;23(8):5063-5088. doi: 10.1007/s10238-023-01207-w. Epub 2023 Oct 7. Clin Exp Med. 2023. PMID: 37804357 Free PMC article.
-
Ataxia Telangiectasia-Mutated Is Activated but Not Required for Productive Autographa californica Multiple Nucleopolyhedrovirus Infection.J Virol. 2022 Nov 23;96(22):e0126922. doi: 10.1128/jvi.01269-22. Epub 2022 Oct 31. J Virol. 2022. PMID: 36314821 Free PMC article.
-
Immunity, virus evolution, and effectiveness of SARS-CoV-2 vaccines.Braz J Med Biol Res. 2021 Mar 15;54(5):e10725. doi: 10.1590/1414-431X202010725. eCollection 2021. Braz J Med Biol Res. 2021. PMID: 33729394 Free PMC article. Review.
References
-
- Hustedt N, Durocher D. 2017. The control of DNA repair by the cell cycle. Nat. Cell Biol. 19:1–9 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
