

Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis
- PMID: 29910178
- PMCID: PMC6094356
- DOI: 10.1016/j.ymthe.2018.05.014
Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis
Abstract
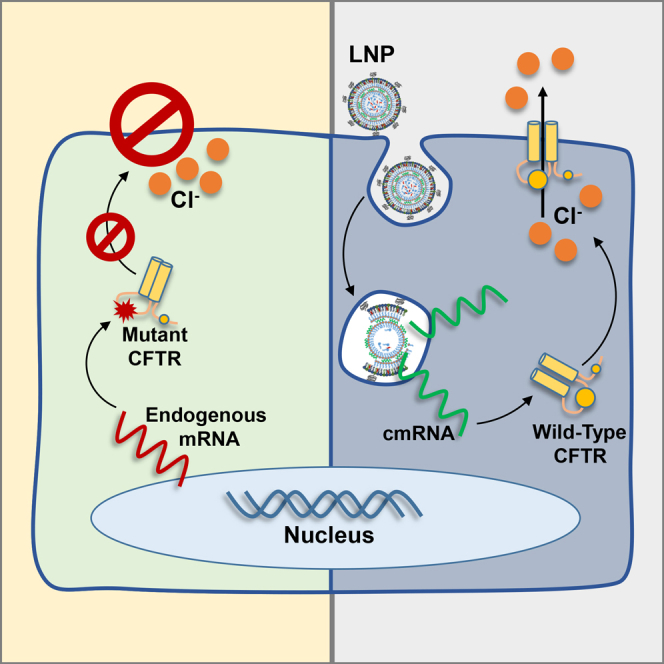
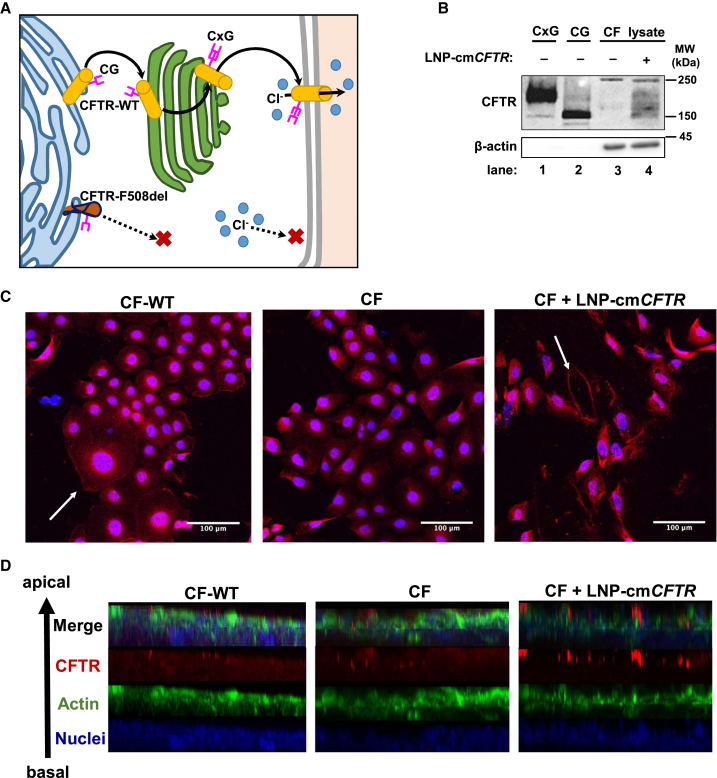
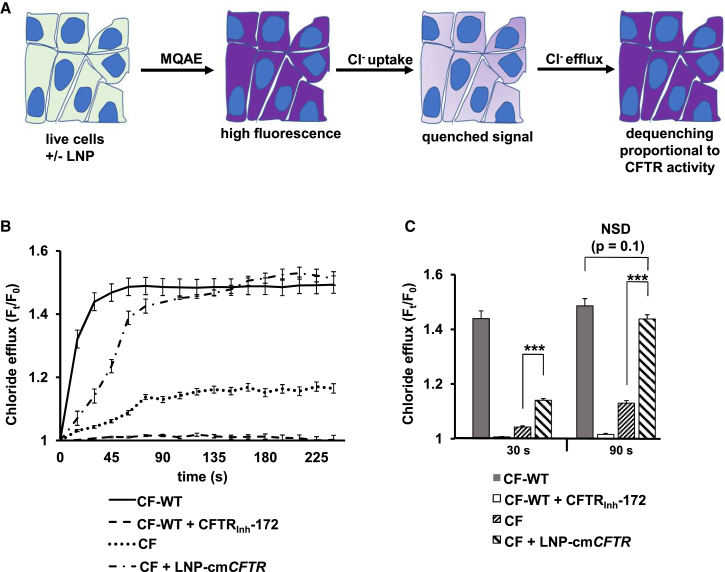
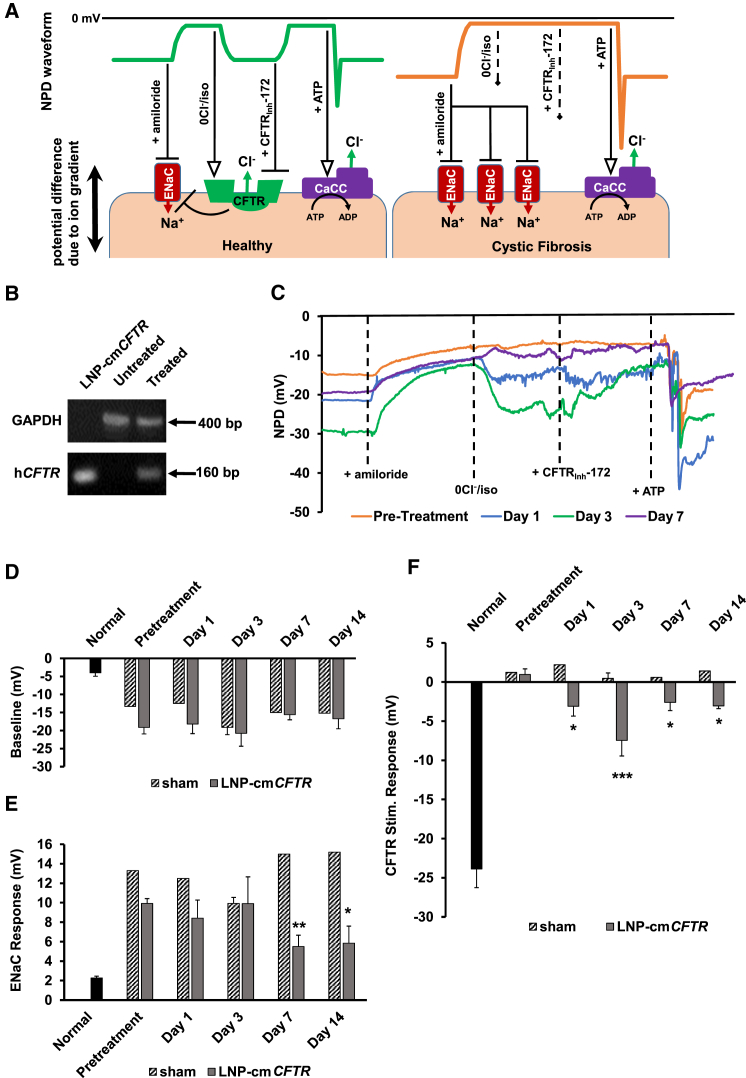
The promise of gene therapy for the treatment of cystic fibrosis has yet to be fully clinically realized despite years of effort toward correcting the underlying genetic defect in the cystic fibrosis transmembrane conductance regulator (CFTR). mRNA therapy via nanoparticle delivery represents a powerful technology for the transfer of genetic material to cells with large, widespread populations, such as airway epithelia. We deployed a clinically relevant lipid-based nanoparticle (LNP) for packaging and delivery of large chemically modified CFTR mRNA (cmCFTR) to patient-derived bronchial epithelial cells, resulting in an increase in membrane-localized CFTR and rescue of its primary function as a chloride channel. Furthermore, nasal application of LNP-cmCFTR restored CFTR-mediated chloride secretion to conductive airway epithelia in CFTR knockout mice for at least 14 days. On day 3 post-transfection, CFTR activity peaked, recovering up to 55% of the net chloride efflux characteristic of healthy mice. This magnitude of response is superior to liposomal CFTR DNA delivery and is comparable with outcomes observed in the currently approved drug ivacaftor. LNP-cmRNA-based systems represent a powerful platform technology for correction of cystic fibrosis and other monogenic disorders.
Keywords: cystic fibrosis; gene therapy; ion transport; mRNA therapeutics; nanoparticles; nasal potential difference.
Copyright © 2018 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy.J Gene Med. 2013 Nov-Dec;15(11-12):414-26. doi: 10.1002/jgm.2748. J Gene Med. 2013. PMID: 24123772
-
A double-blind, placebo controlled, dose ranging study to evaluate the safety and biological efficacy of the lipid-DNA complex GR213487B in the nasal epithelium of adult patients with cystic fibrosis.Hum Gene Ther. 1998 Jan 20;9(2):249-69. doi: 10.1089/hum.1998.9.2-249. Hum Gene Ther. 1998. PMID: 9472784 Clinical Trial.
-
Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells.Hum Gene Ther. 2005 Sep;16(9):1116-23. doi: 10.1089/hum.2005.16.1116. Hum Gene Ther. 2005. PMID: 16149910
-
Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease.Cochrane Database Syst Rev. 2007 Apr 18;(2):CD005599. doi: 10.1002/14651858.CD005599.pub2. Cochrane Database Syst Rev. 2007. Update in: Cochrane Database Syst Rev. 2012 Oct 17;10:CD005599. doi: 10.1002/14651858.CD005599.pub3. PMID: 17443603 Updated. Review.
-
Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease.Cochrane Database Syst Rev. 2013 Nov 26;(11):CD005599. doi: 10.1002/14651858.CD005599.pub4. Cochrane Database Syst Rev. 2013. Update in: Cochrane Database Syst Rev. 2016 Jun 17;(6):CD005599. doi: 10.1002/14651858.CD005599.pub5. PMID: 24282073 Updated. Review.
Cited by
-
Therapeutic RNA Delivery for COVID and Other Diseases.Adv Healthc Mater. 2021 Aug;10(15):e2002022. doi: 10.1002/adhm.202002022. Epub 2021 Mar 4. Adv Healthc Mater. 2021. PMID: 33661555 Free PMC article. Review.
-
Evaluation of the efficacy of cystinosin supplementation through CTNS mRNA delivery in experimental models for cystinosis.Sci Rep. 2023 Nov 28;13(1):20961. doi: 10.1038/s41598-023-47085-w. Sci Rep. 2023. PMID: 38016974 Free PMC article.
-
Harnessing lipid nanoparticles for efficient CRISPR delivery.Biomater Sci. 2021 Sep 14;9(18):6001-6011. doi: 10.1039/d1bm00537e. Biomater Sci. 2021. PMID: 34115079 Free PMC article. Review.
-
Lipid-based nucleic acid therapeutics with in vivo efficacy.Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2023 Mar;15(2):e1856. doi: 10.1002/wnan.1856. Epub 2022 Sep 30. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2023. PMID: 36180107 Free PMC article. Review.
-
Capsaicin-Loaded Chitosan Nanocapsules for wtCFTR-mRNA Delivery to a Cystic Fibrosis Cell Line.Biomedicines. 2020 Sep 20;8(9):364. doi: 10.3390/biomedicines8090364. Biomedicines. 2020. PMID: 32962254 Free PMC article.
References
-
- Cystic Fibrosis Foundation (2018) About Cystic Fibrosis. https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/.
-
- Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Welsh M.J. Abnormal regulation of ion channels in cystic fibrosis epithelia. FASEB J. 1990;4:2718–2725. - PubMed
-
- Chu C.S., Trapnell B.C., Curristin S., Cutting G.R., Crystal R.G. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 1993;3:151–156. - PubMed
-
- Welsh M.J., Anderson M.P., Rich D.P., Berger H.A., Denning G.M., Ostedgaard L.S., Sheppard D.N., Cheng S.H., Gregory R.J., Smith A.E. Cystic fibrosis transmembrane conductance regulator: a chloride channel with novel regulation. Neuron. 1992;8:821–829. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
