

Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology
- PMID: 29873596
- PMCID: PMC6335102
- DOI: 10.1152/physrev.00038.2017
Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology
Abstract
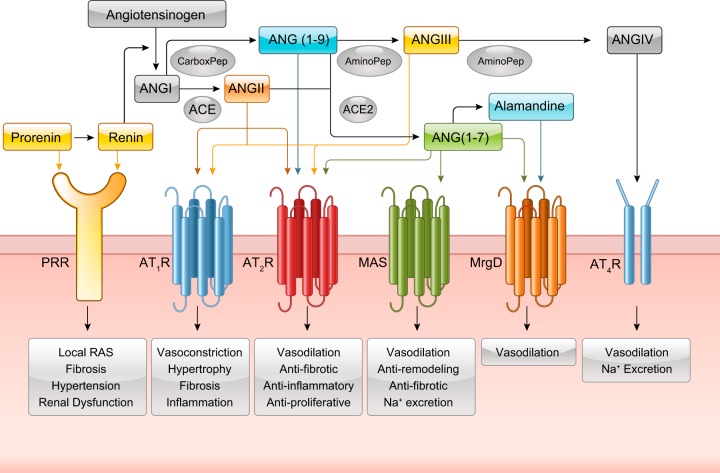
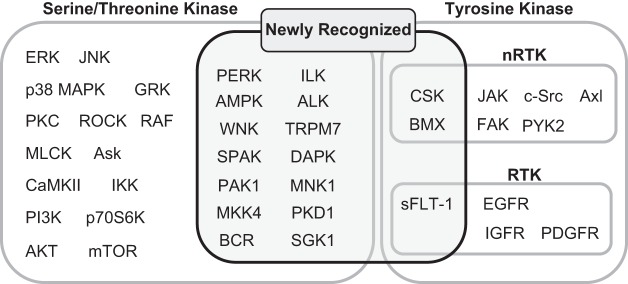
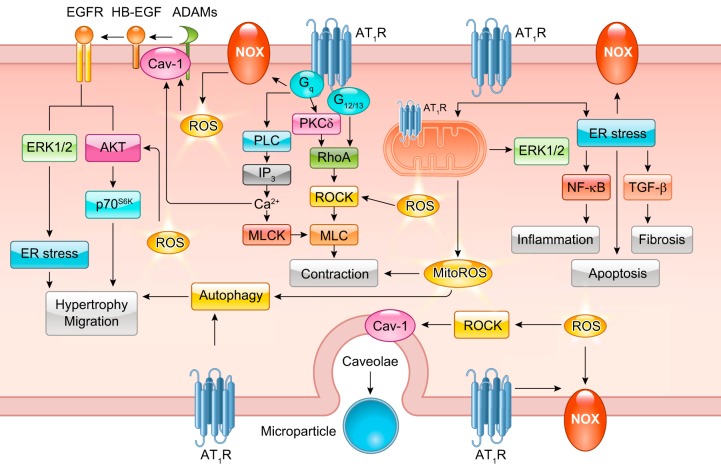
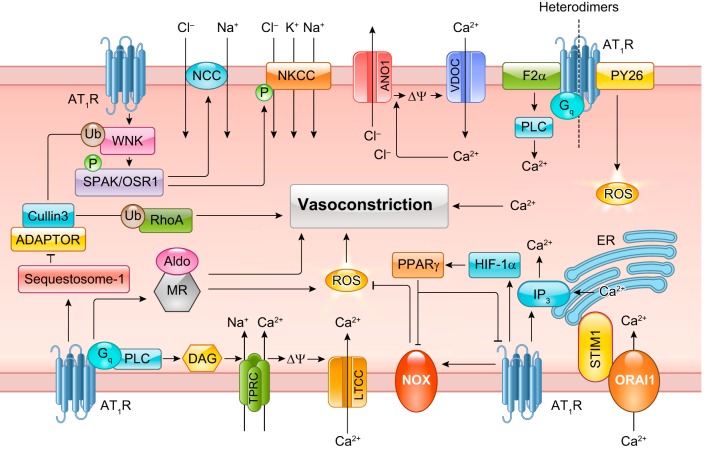
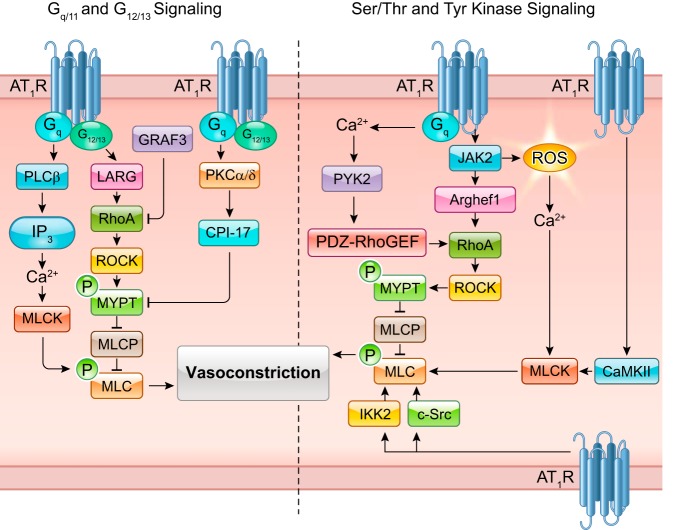
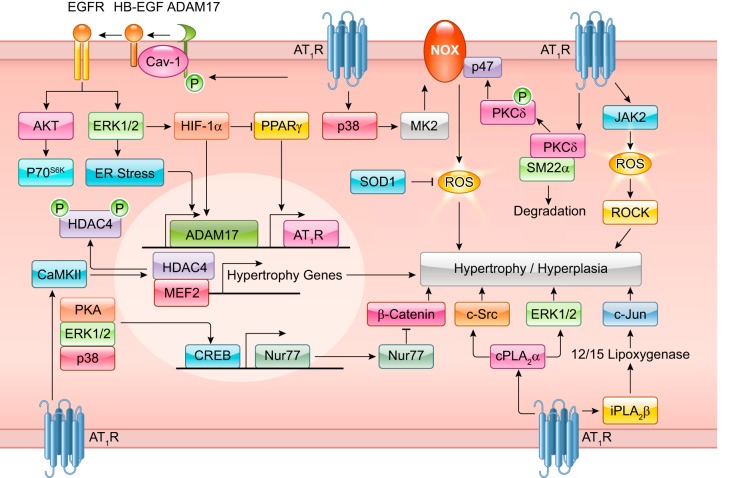
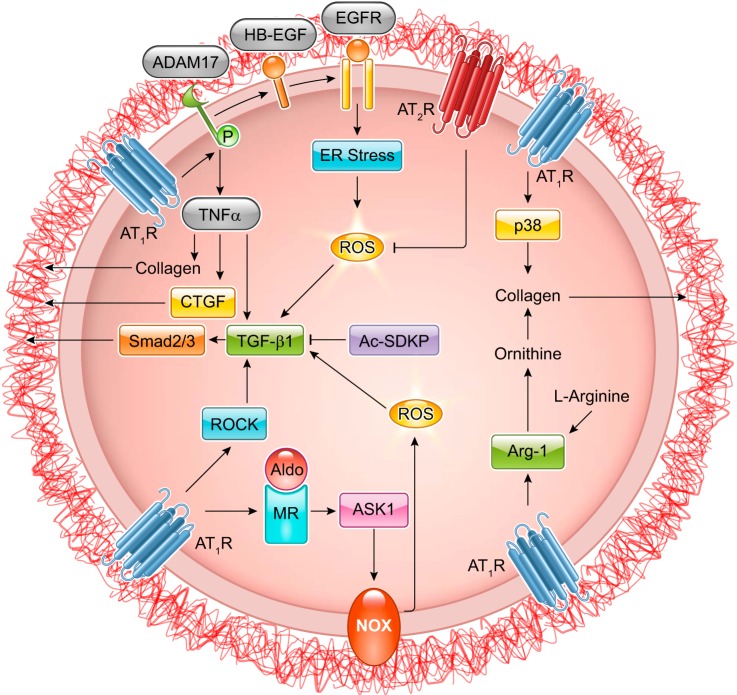
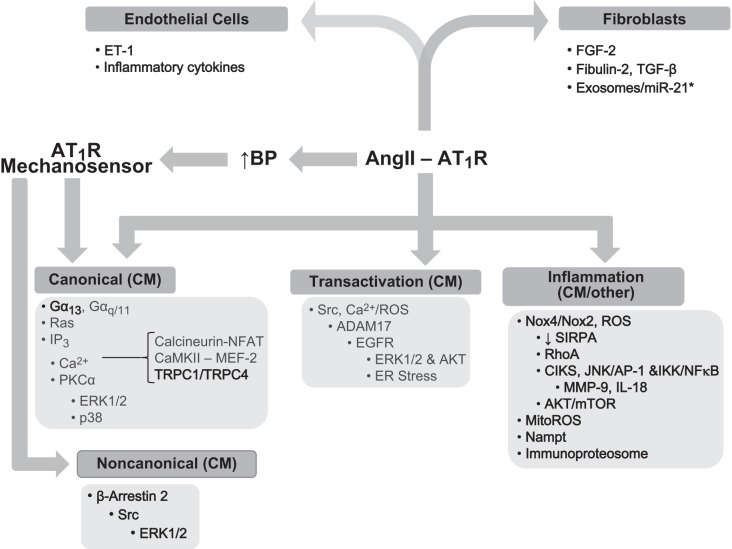
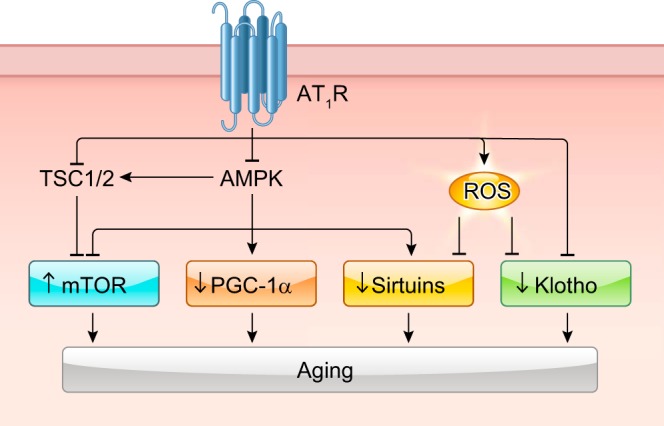
The renin-angiotensin-aldosterone system plays crucial roles in cardiovascular physiology and pathophysiology. However, many of the signaling mechanisms have been unclear. The angiotensin II (ANG II) type 1 receptor (AT1R) is believed to mediate most functions of ANG II in the system. AT1R utilizes various signal transduction cascades causing hypertension, cardiovascular remodeling, and end organ damage. Moreover, functional cross-talk between AT1R signaling pathways and other signaling pathways have been recognized. Accumulating evidence reveals the complexity of ANG II signal transduction in pathophysiology of the vasculature, heart, kidney, and brain, as well as several pathophysiological features, including inflammation, metabolic dysfunction, and aging. In this review, we provide a comprehensive update of the ANG II receptor signaling events and their functional significances for potential translation into therapeutic strategies. AT1R remains central to the system in mediating physiological and pathophysiological functions of ANG II, and participation of specific signaling pathways becomes much clearer. There are still certain limitations and many controversies, and several noteworthy new concepts require further support. However, it is expected that rigorous translational research of the ANG II signaling pathways including those in large animals and humans will contribute to establishing effective new therapies against various diseases.
Figures
Similar articles
-
New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone.J Mol Med (Berl). 2008 Jun;86(6):673-8. doi: 10.1007/s00109-008-0323-5. Epub 2008 Mar 27. J Mol Med (Berl). 2008. PMID: 18368382 Review.
-
Angiotensin II type 2 receptor (AT2R) in renal and cardiovascular disease.Clin Sci (Lond). 2016 Aug 1;130(15):1307-26. doi: 10.1042/CS20160243. Clin Sci (Lond). 2016. PMID: 27358027 Review.
-
Renal denervation attenuates aldosterone expression and associated cardiovascular pathophysiology in angiotensin II-induced hypertension.Oncotarget. 2016 Oct 18;7(42):67828-67840. doi: 10.18632/oncotarget.12182. Oncotarget. 2016. PMID: 27661131 Free PMC article.
-
ACE2 and vasoactive peptides: novel players in cardiovascular/renal remodeling and hypertension.Ther Adv Cardiovasc Dis. 2015 Aug;9(4):217-37. doi: 10.1177/1753944715597623. Epub 2015 Aug 13. Ther Adv Cardiovasc Dis. 2015. PMID: 26275770 Review.
-
Cross-talk between inflammation and angiotensin II: studies based on direct transfection of cardiomyocytes with AT1R and AT2R cDNA.Exp Biol Med (Maywood). 2012 Dec;237(12):1394-401. doi: 10.1258/ebm.2012.012212. Exp Biol Med (Maywood). 2012. PMID: 23354398
Cited by
-
Organ crosstalk and dysfunction in sepsis.Ann Intensive Care. 2024 Sep 19;14(1):147. doi: 10.1186/s13613-024-01377-0. Ann Intensive Care. 2024. PMID: 39298039 Free PMC article. Review.
-
Losartan attenuates upstream vasculopathy in a modified piglet model of pulmonary vein stenosis: contribution of the Hippo pathway.Ann Transl Med. 2022 Nov;10(21):1153. doi: 10.21037/atm-22-2621. Ann Transl Med. 2022. PMID: 36467357 Free PMC article.
-
Perindopril and losartan affect ACE-2 and IL- 6 expression in obese rat model.Narra J. 2024 Aug;4(2):e681. doi: 10.52225/narra.v4i2.681. Epub 2024 Jul 15. Narra J. 2024. PMID: 39280311 Free PMC article.
-
Ibrutinib-induced pulmonary angiotensin-converting enzyme activation promotes atrial fibrillation in rats.iScience. 2024 Jan 17;27(2):108926. doi: 10.1016/j.isci.2024.108926. eCollection 2024 Feb 16. iScience. 2024. PMID: 38357670 Free PMC article.
-
Aliskiren Attenuates the Inflammatory Response and Wound Healing Process in Diabetic Mice With Periodontal Disease.Front Pharmacol. 2019 Jul 4;10:708. doi: 10.3389/fphar.2019.00708. eCollection 2019. Front Pharmacol. 2019. PMID: 31333451 Free PMC article.
References
-
- Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA 108: 14849–14854, 2011. doi:10.1073/pnas.1101507108. - DOI - PMC - PubMed
-
- Abbas M, Jesel L, Auger C, Amoura L, Messas N, Manin G, Rumig C, León-González AJ, Ribeiro TP, Silva GC, Abou-Merhi R, Hamade E, Hecker M, Georg Y, Chakfe N, Ohlmann P, Schini-Kerth VB, Toti F, Morel O. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 135: 280–296, 2017. doi:10.1161/CIRCULATIONAHA.116.017513. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous