

Myotonic Dystrophy and Developmental Regulation of RNA Processing
- PMID: 29687899
- PMCID: PMC11323716
- DOI: 10.1002/cphy.c170002
Myotonic Dystrophy and Developmental Regulation of RNA Processing
Abstract
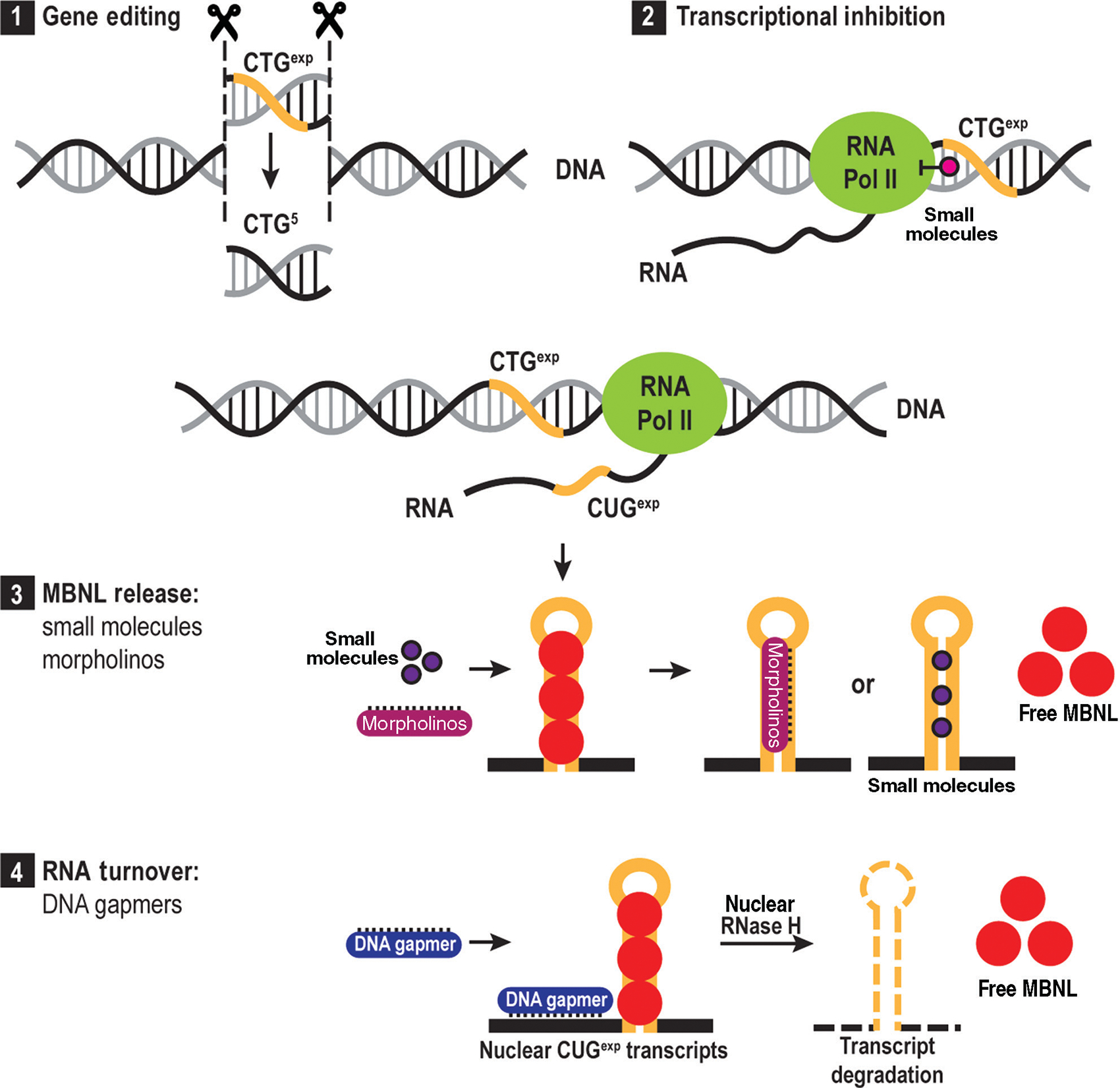
Myotonic dystrophy (DM) is a multisystemic disorder caused by microsatellite expansion mutations in two unrelated genes leading to similar, yet distinct, diseases. DM disease presentation is highly variable and distinguished by differences in age-of-onset and symptom severity. In the most severe form, DM presents with congenital onset and profound developmental defects. At the molecular level, DM pathogenesis is characterized by a toxic RNA gain-of-function mechanism that involves the transcription of noncoding microsatellite expansions. These mutant RNAs disrupt key cellular pathways, including RNA processing, localization, and translation. In DM, these toxic RNA effects are predominantly mediated through the modulation of the muscleblind-like and CUGBP and ETR-3-like factor families of RNA binding proteins (RBPs). Dysfunction of these RBPs results in widespread RNA processing defects culminating in the expression of developmentally inappropriate protein isoforms in adult tissues. The tissue that is the focus of this review, skeletal muscle, is particularly sensitive to mutant RNA-responsive perturbations, as patients display a variety of developmental, structural, and functional defects in muscle. Here, we provide a comprehensive overview of DM1 and DM2 clinical presentation and pathology as well as the underlying cellular and molecular defects associated with DM disease onset and progression. Additionally, fundamental aspects of skeletal muscle development altered in DM are highlighted together with ongoing and potential therapeutic avenues to treat this muscular dystrophy. © 2018 American Physiological Society. Compr Physiol 8:509-553, 2018.
Copyright © 2018 American Physiological Society. All rights reserved.
Figures

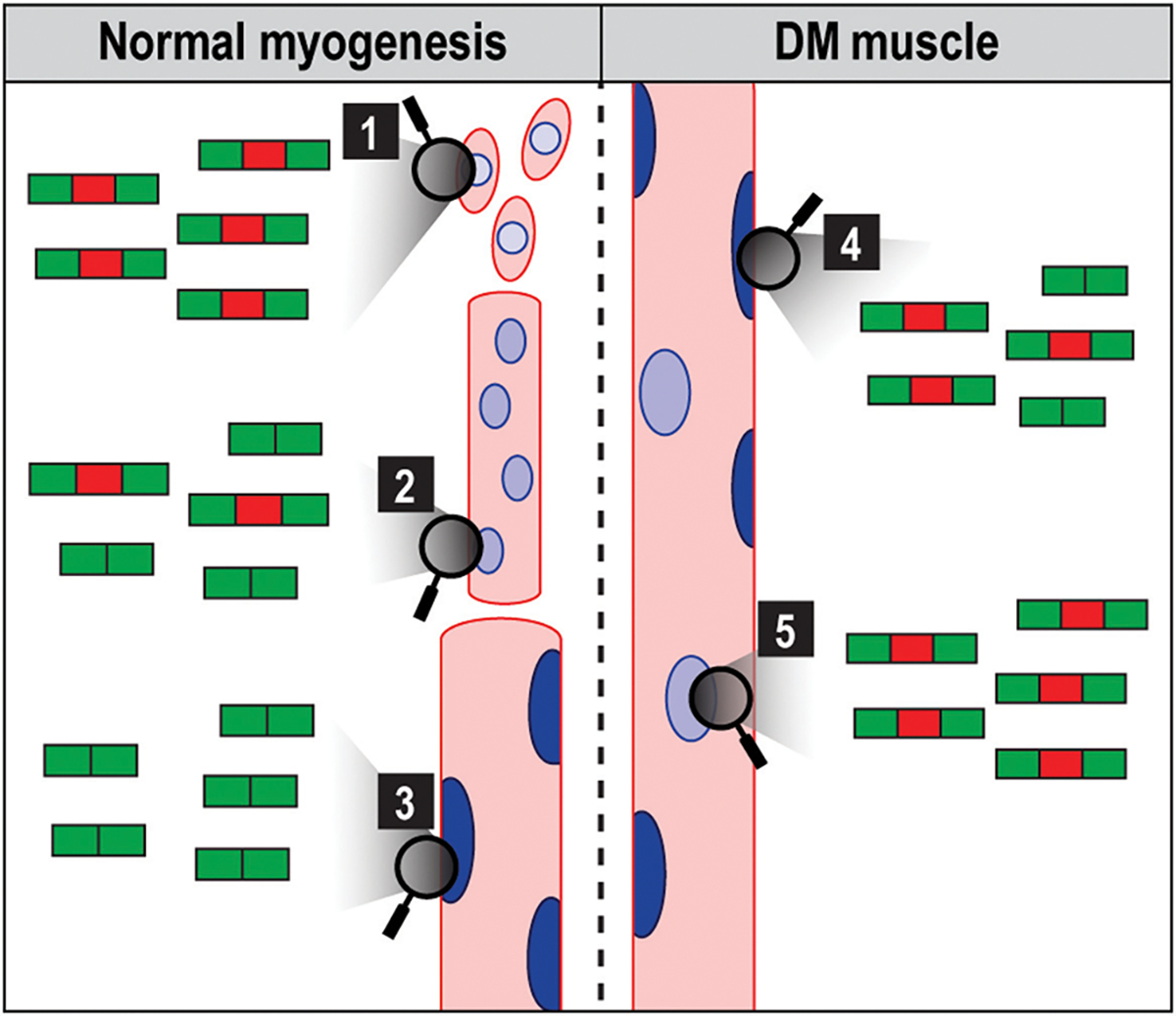
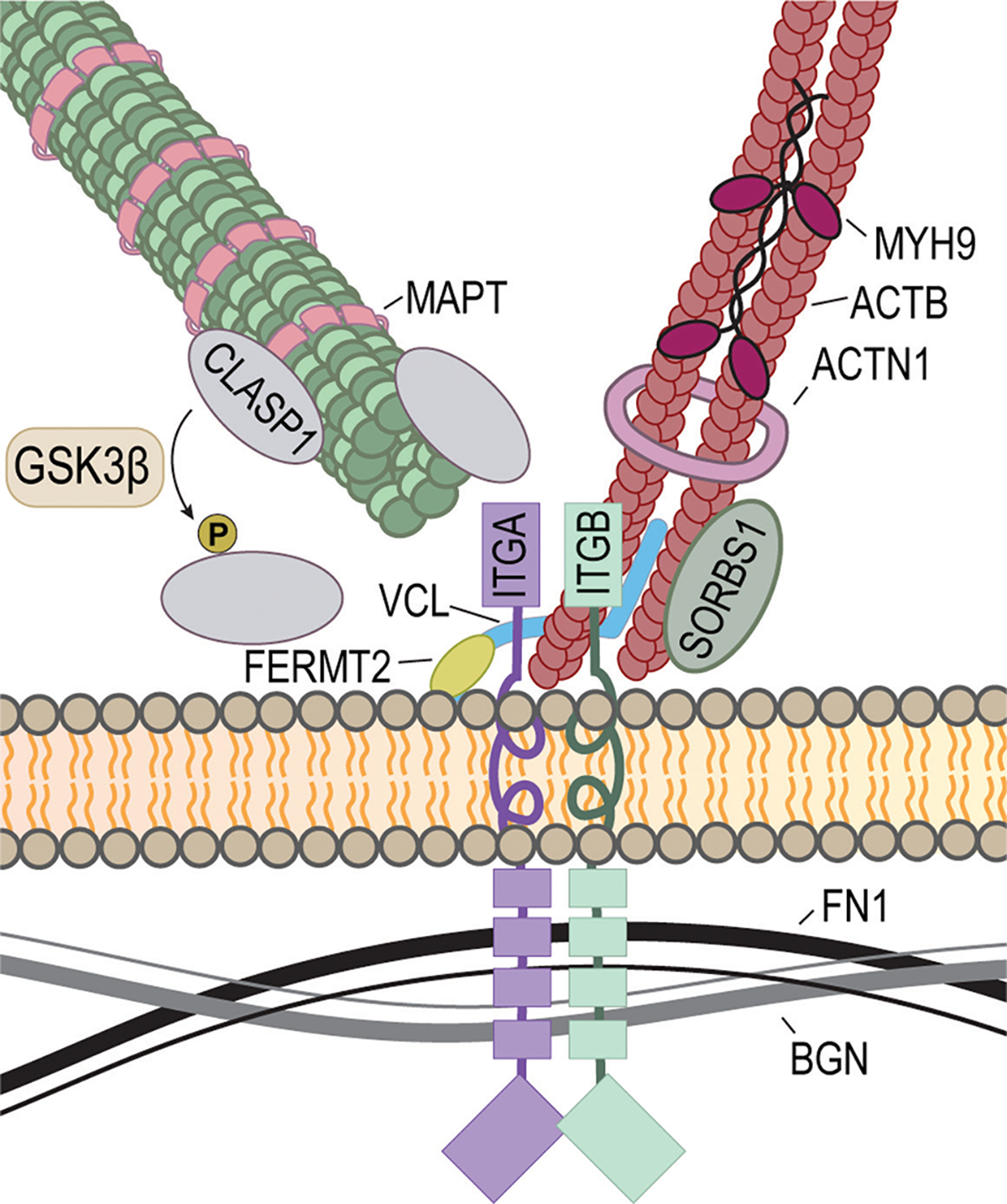
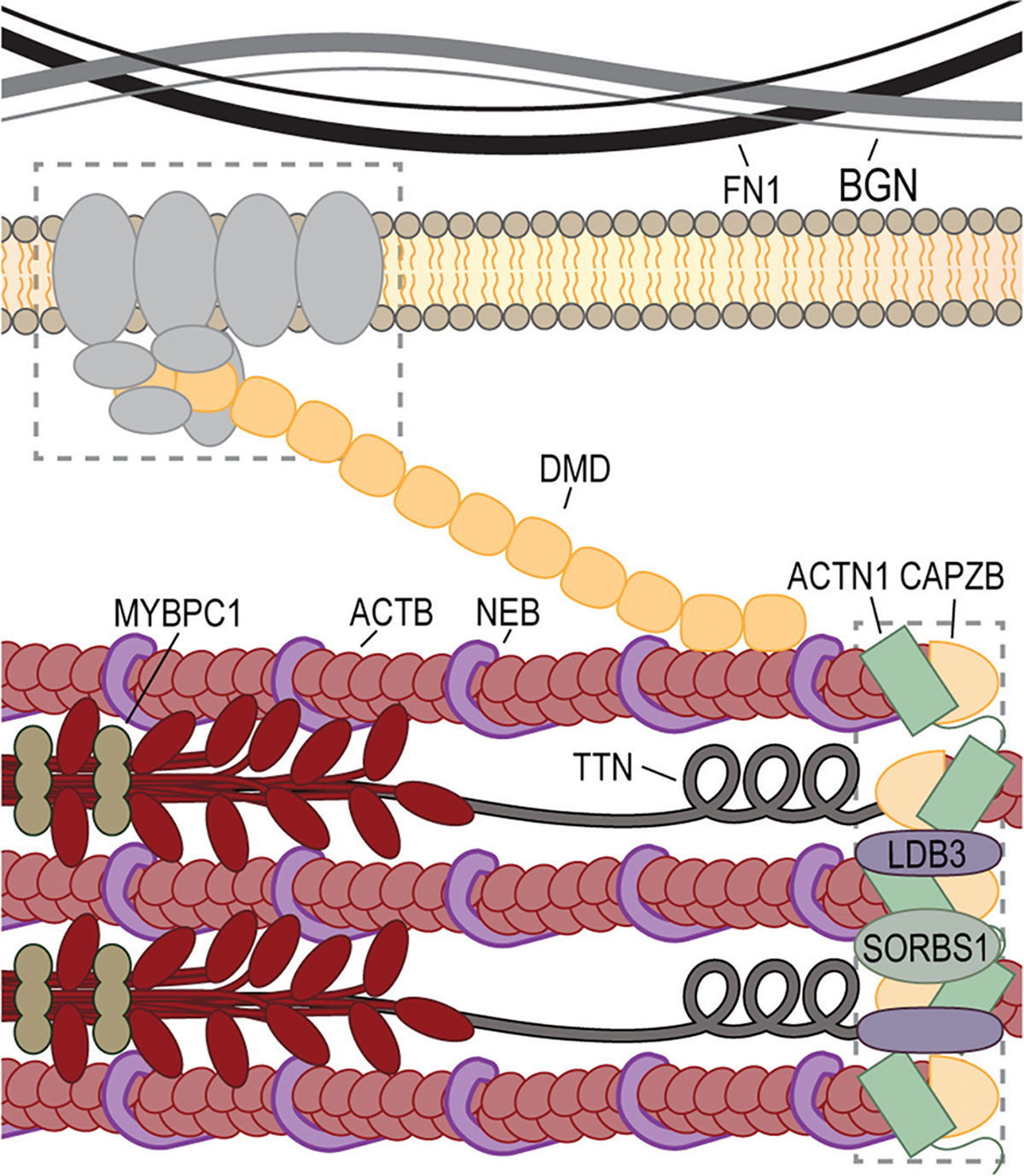
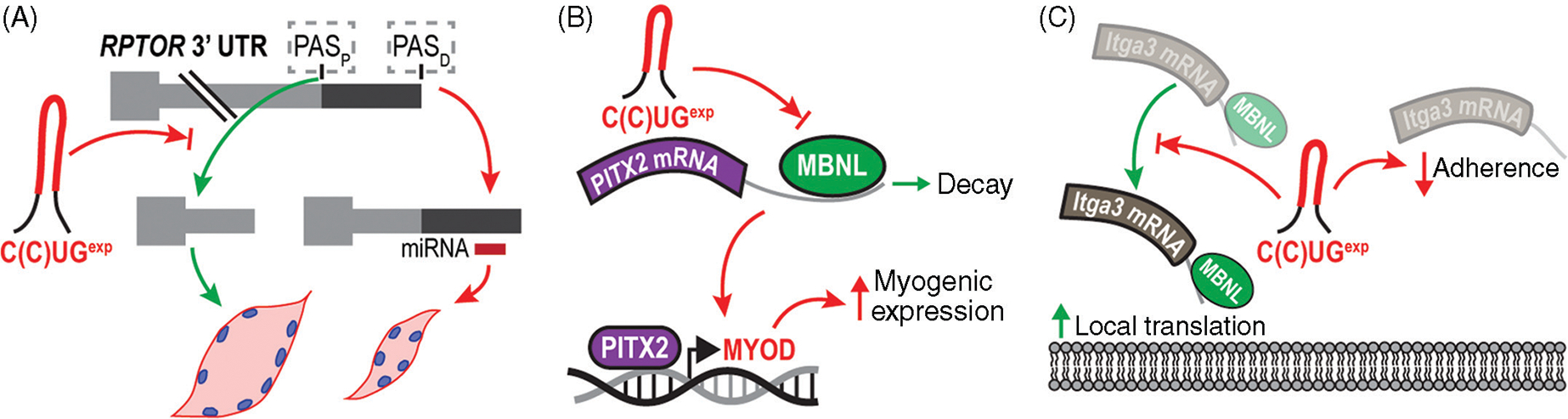
Similar articles
-
Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy.Genes Dev. 2017 Jun 1;31(11):1122-1133. doi: 10.1101/gad.300590.117. Epub 2017 Jul 11. Genes Dev. 2017. PMID: 28698297 Free PMC article.
-
Flies deficient in Muscleblind protein model features of myotonic dystrophy with altered splice forms of Z-band associated transcripts.Hum Genet. 2006 Nov;120(4):487-99. doi: 10.1007/s00439-006-0228-8. Epub 2006 Aug 23. Hum Genet. 2006. PMID: 16927100
-
Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes.JCI Insight. 2019 Mar 21;4(6):e122686. doi: 10.1172/jci.insight.122686. eCollection 2019 Mar 21. JCI Insight. 2019. PMID: 30730308 Free PMC article.
-
Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies.Acta Myol. 2013 Dec;32(3):154-65. Acta Myol. 2013. PMID: 24803843 Free PMC article. Review.
-
Myotonic dystrophy: RNA pathogenesis comes into focus.Am J Hum Genet. 2004 May;74(5):793-804. doi: 10.1086/383590. Epub 2004 Apr 2. Am J Hum Genet. 2004. PMID: 15065017 Free PMC article. Review.
Cited by
-
Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies.Int J Mol Sci. 2022 Jan 27;23(3):1441. doi: 10.3390/ijms23031441. Int J Mol Sci. 2022. PMID: 35163365 Free PMC article. Review.
-
Disrupting the Molecular Pathway in Myotonic Dystrophy.Int J Mol Sci. 2021 Dec 8;22(24):13225. doi: 10.3390/ijms222413225. Int J Mol Sci. 2021. PMID: 34948025 Free PMC article. Review.
-
MBNL splicing factors regulate the microtranscriptome of skeletal muscles.Nucleic Acids Res. 2024 Oct 28;52(19):12055-12073. doi: 10.1093/nar/gkae774. Nucleic Acids Res. 2024. PMID: 39258536 Free PMC article.
-
A framework for translating tauopathy therapeutics: Drug discovery to clinical trials.Alzheimers Dement. 2024 Nov;20(11):8129-8152. doi: 10.1002/alz.14250. Epub 2024 Sep 24. Alzheimers Dement. 2024. PMID: 39316411 Free PMC article. Review.
-
Specific DMPK-promoter targeting by CRISPRi reverses myotonic dystrophy type 1-associated defects in patient muscle cells.Mol Ther Nucleic Acids. 2023 May 13;32:857-871. doi: 10.1016/j.omtn.2023.05.007. eCollection 2023 Jun 13. Mol Ther Nucleic Acids. 2023. PMID: 37273786 Free PMC article.
References
-
- Abmayr SM, Balagopalan L, Galletta BJ, Hong SJ. Cell and molecular biology of myoblast fusion. Int Rev Cytol 225: 33–89, 2003. - PubMed
-
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104: 923–935, 2001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
