

Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3
- PMID: 29626115
- PMCID: PMC5940261
- DOI: 10.1084/jem.20171417
Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3
Abstract
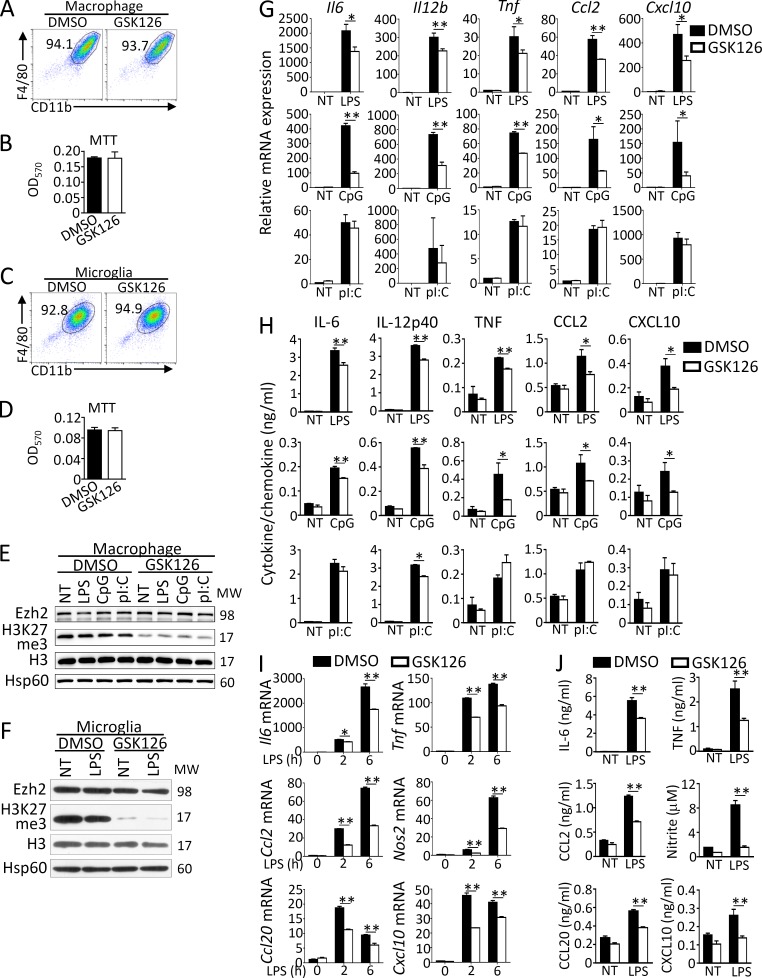
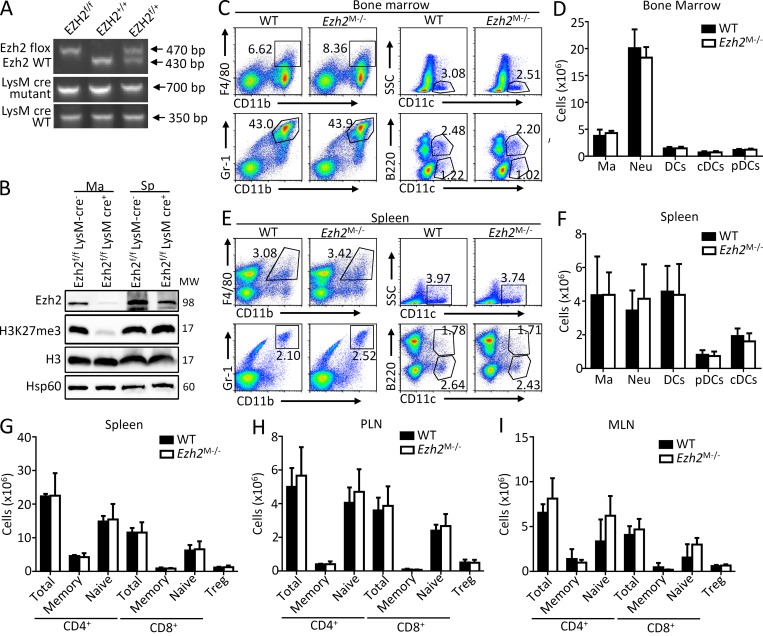
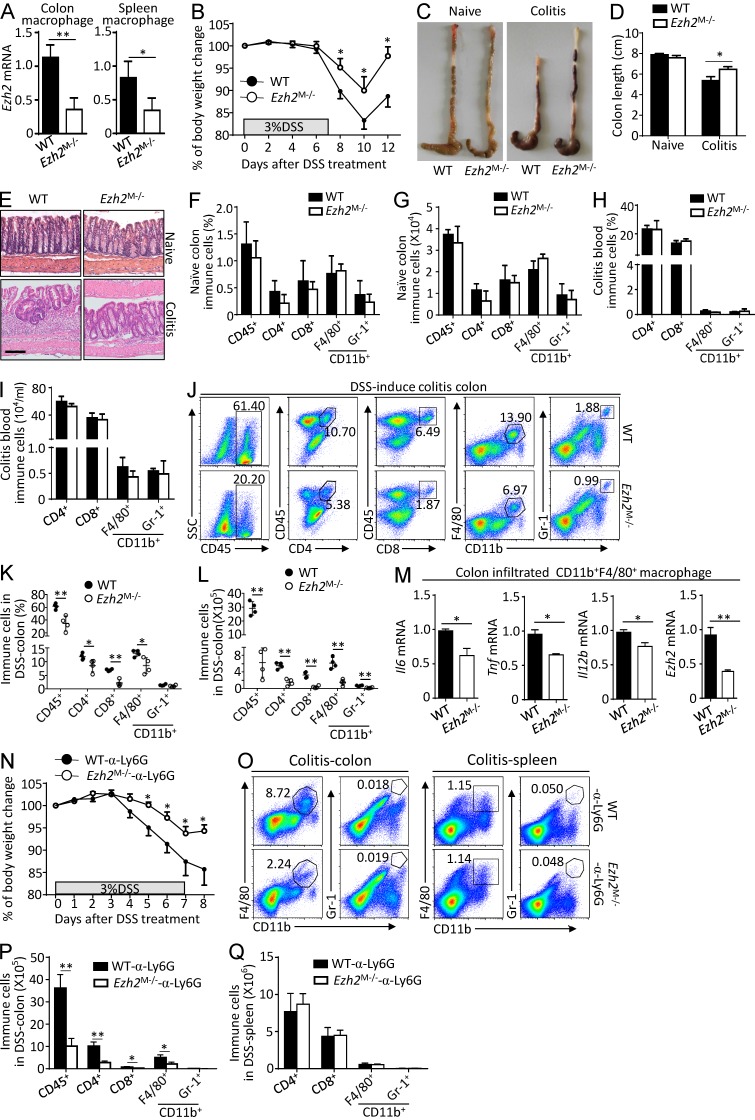
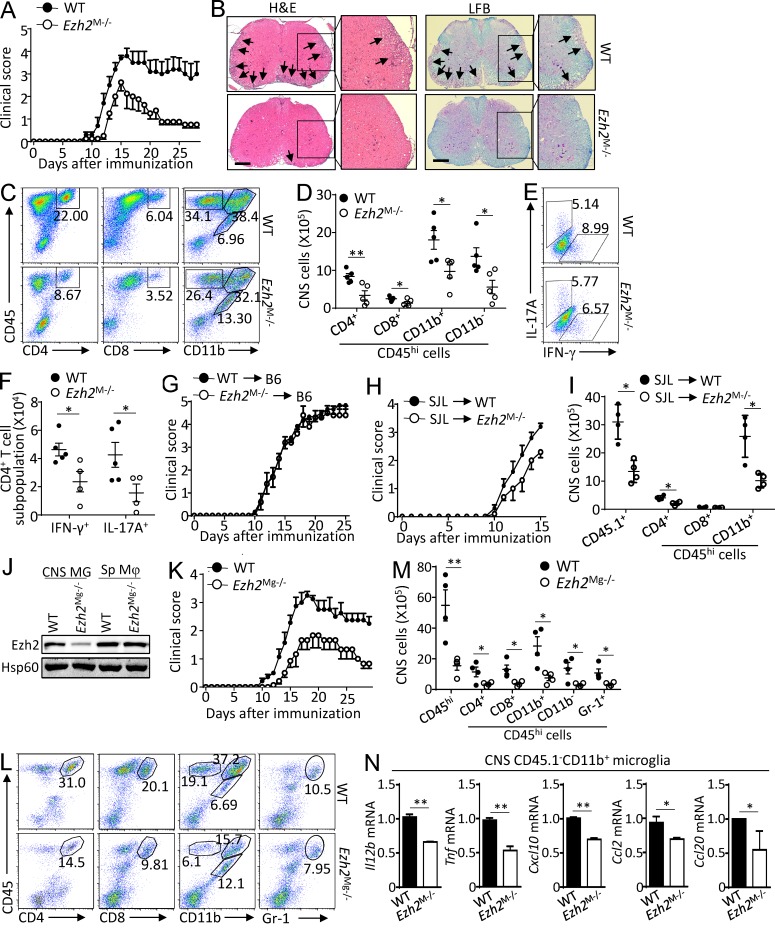
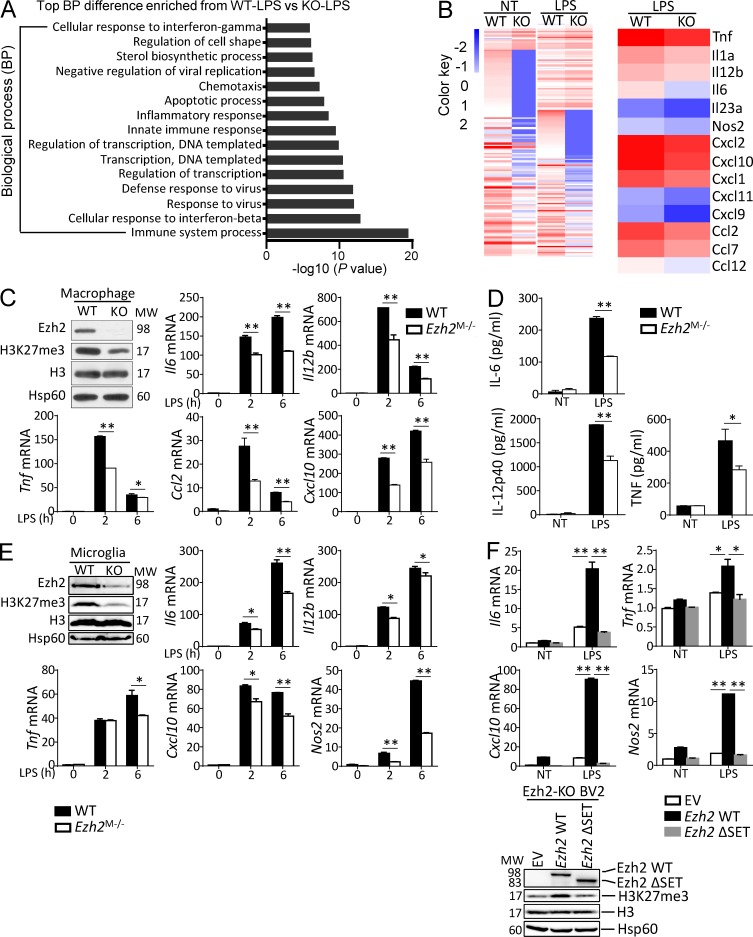
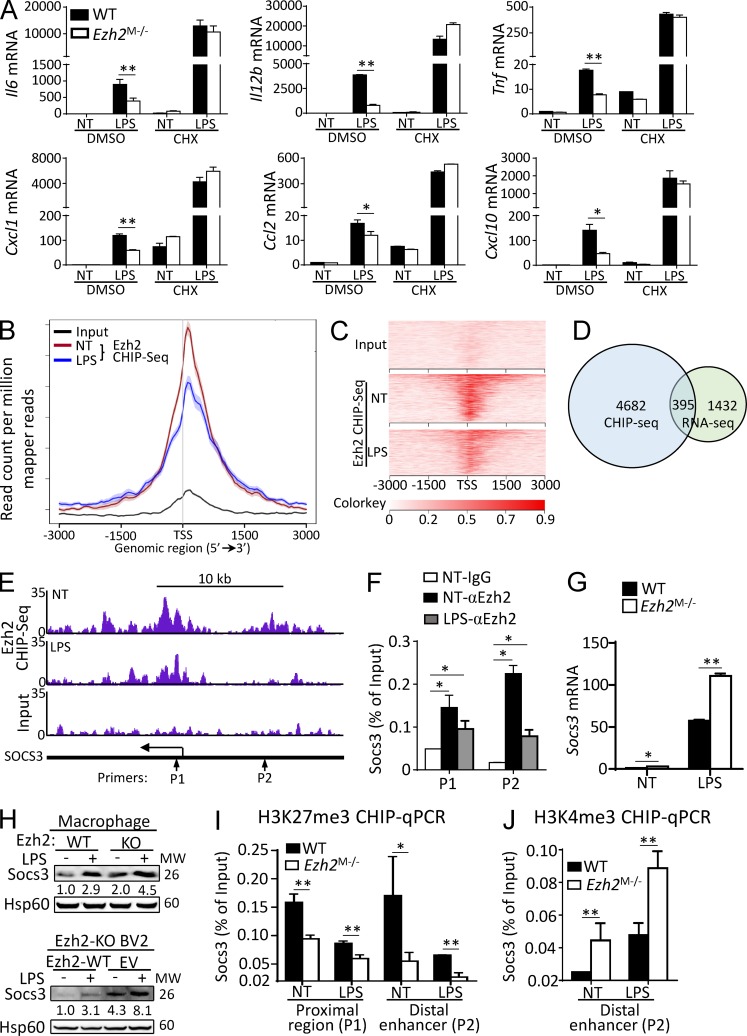
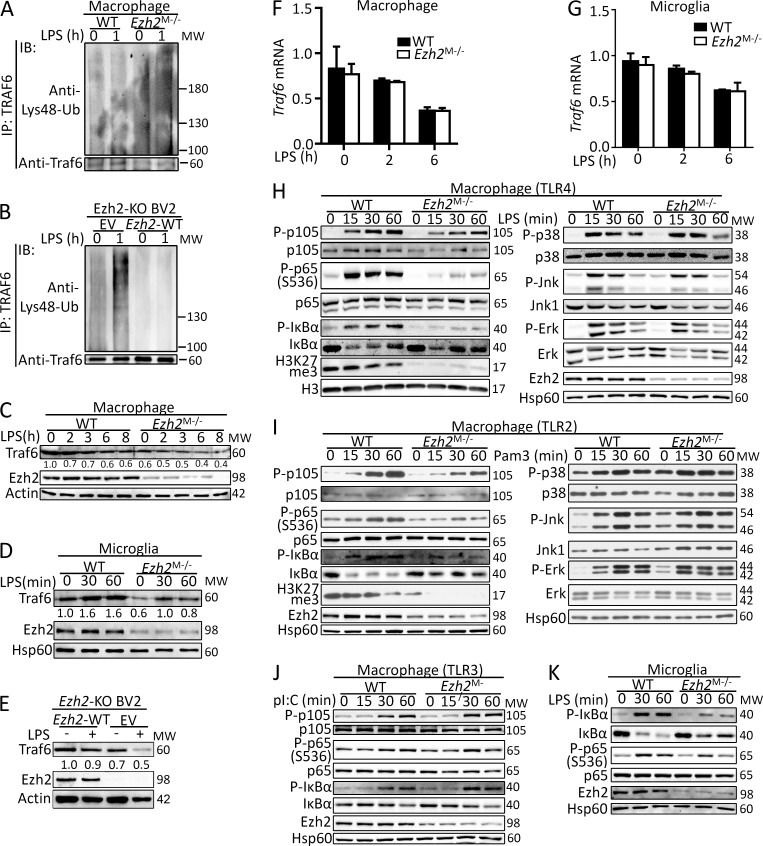
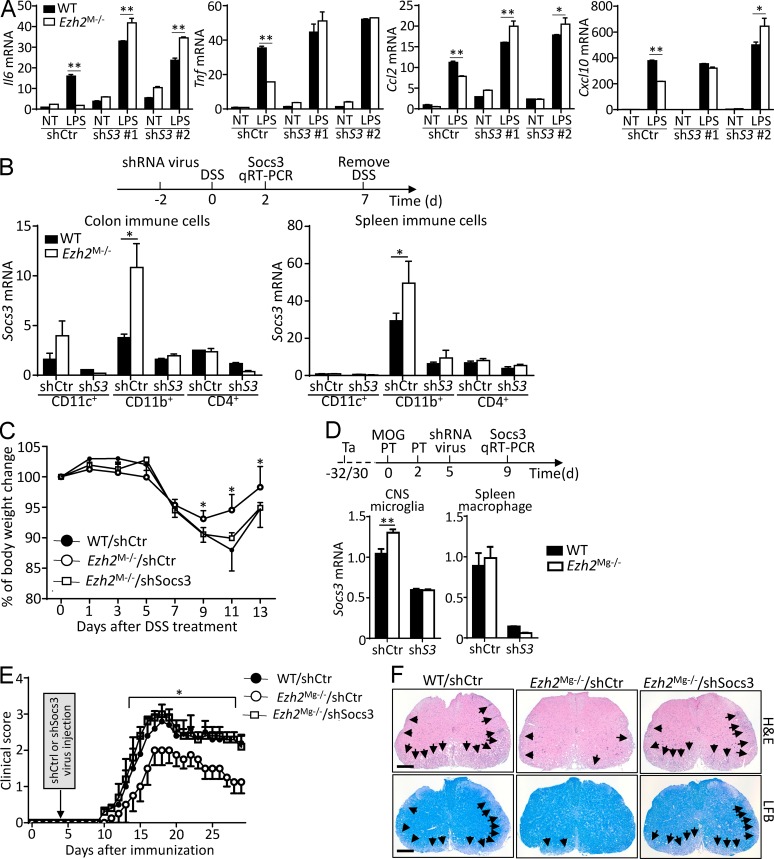
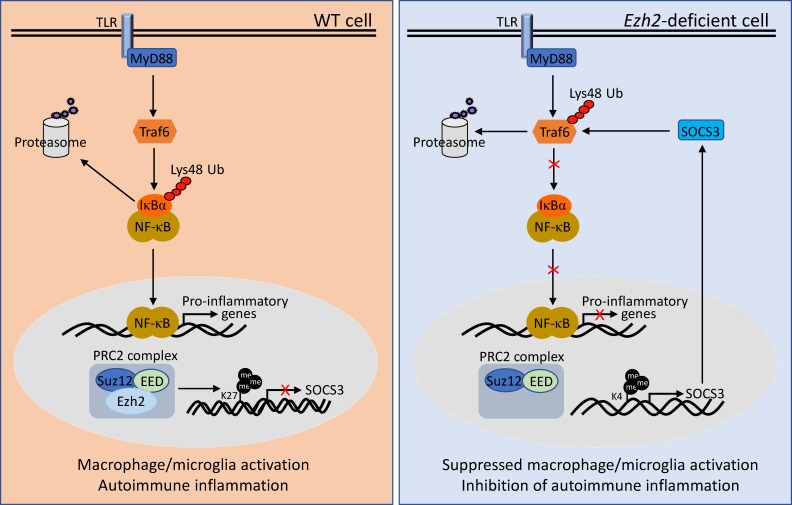
Histone 3 Lys27 (H3K27) trimethyltransferase Ezh2 is implicated in the pathogenesis of autoimmune inflammation. Nevertheless, the role of Ezh2 in macrophage/microglial activation remains to be defined. In this study, we identified that macrophage/microglial H3K27me3 or Ezh2, rather than functioning as a repressor, mediates toll-like receptor (TLR)-induced proinflammatory gene expression, and therefore Ezh2 depletion diminishes macrophage/microglial activation and attenuates the autoimmune inflammation in dextran sulfate sodium-induced colitis and experimental autoimmune encephalomyelitis. Mechanistic characterizations indicated that Ezh2 deficiency directly stimulates suppressor of cytokine signaling 3 (Socs3) expression and therefore enhances the Lys48-linked ubiquitination and degradation of tumor necrosis factor receptor-associated factor 6. As a consequence, TLR-induced MyD88-dependent nuclear factor κB activation and the expression of proinflammatory genes in macrophages/microglia are compromised in the absence of Ezh2. The functional dependence of Ezh2 for Socs3 is further illustrated by the rescue experiments in which silencing of Socs3 restores macrophage activation and rescues autoimmune inflammation in macrophage/microglial Ezh2-deficient mice. Together, these findings establish Ezh2 as a macrophage lineage-specific mediator of autoimmune inflammation and highlight a previously unknown mechanism of Ezh2 function.
© 2018 Zhang et al.
Figures
Comment in
-
Repressing the repressor: Ezh2 mediates macrophage activation.J Exp Med. 2018 May 7;215(5):1269-1271. doi: 10.1084/jem.20180479. Epub 2018 Apr 24. J Exp Med. 2018. PMID: 29691302 Free PMC article.
Similar articles
-
Inhibition of EZH2 (Enhancer of Zeste Homolog 2) Attenuates Neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-κB (Trimethylation of Histone 3 Lysine 27/Suppressor of Cytokine Signaling 3/Tumor Necrosis Factor Receptor Family 6/Nuclear Factor-κB) in a Rat Model of Subarachnoid Hemorrhage.Stroke. 2020 Nov;51(11):3320-3331. doi: 10.1161/STROKEAHA.120.029951. Epub 2020 Sep 16. Stroke. 2020. PMID: 32933418 Free PMC article.
-
Calcitonin gene-related peptide regulates spinal microglial activation through the histone H3 lysine 27 trimethylation via enhancer of zeste homolog-2 in rats with neuropathic pain.J Neuroinflammation. 2021 May 21;18(1):117. doi: 10.1186/s12974-021-02168-1. J Neuroinflammation. 2021. PMID: 34020664 Free PMC article.
-
Enhancer of zeste homolog 2-catalysed H3K27 trimethylation plays a key role in acute-on-chronic liver failure via TNF-mediated pathway.Cell Death Dis. 2018 May 22;9(6):590. doi: 10.1038/s41419-018-0670-2. Cell Death Dis. 2018. PMID: 29789597 Free PMC article.
-
EZH2 in normal hematopoiesis and hematological malignancies.Oncotarget. 2016 Jan 19;7(3):2284-96. doi: 10.18632/oncotarget.6198. Oncotarget. 2016. PMID: 26497210 Free PMC article. Review.
-
Emerging role of EZH2 in rheumatic diseases: A comprehensive review.Int J Rheum Dis. 2022 Nov;25(11):1230-1238. doi: 10.1111/1756-185X.14416. Epub 2022 Aug 7. Int J Rheum Dis. 2022. PMID: 35933601 Review.
Cited by
-
Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality.ESC Heart Fail. 2021 Jun;8(3):1873-1884. doi: 10.1002/ehf2.13297. Epub 2021 Mar 28. ESC Heart Fail. 2021. PMID: 33779075 Free PMC article.
-
Inhibition of EZH2 Causes Retrotransposon Derepression and Immune Activation in Porcine Lung Alveolar Macrophages.Int J Mol Sci. 2023 Jan 25;24(3):2394. doi: 10.3390/ijms24032394. Int J Mol Sci. 2023. PMID: 36768720 Free PMC article.
-
Transition to a mesenchymal state in neuroblastoma confers resistance to anti-GD2 antibody via reduced expression of ST8SIA1.Nat Cancer. 2022 Aug;3(8):976-993. doi: 10.1038/s43018-022-00405-x. Epub 2022 Jul 11. Nat Cancer. 2022. PMID: 35817829 Free PMC article.
-
Regulation of microglial activation in stroke in aged mice: a translational study.Aging (Albany NY). 2022 Aug 12;14(15):6047-6065. doi: 10.18632/aging.204216. Epub 2022 Aug 12. Aging (Albany NY). 2022. PMID: 35963621 Free PMC article.
-
The Regulatory Roles of Ezh2 in Response to Lipopolysaccharide (LPS) in Macrophages and Mice with Conditional Ezh2 Deletion with LysM-Cre System.Int J Mol Sci. 2023 Mar 10;24(6):5363. doi: 10.3390/ijms24065363. Int J Mol Sci. 2023. PMID: 36982437 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
