

Cryo-EM structure of the polycystic kidney disease-like channel PKD2L1
- PMID: 29567962
- PMCID: PMC5864754
- DOI: 10.1038/s41467-018-03606-0
Cryo-EM structure of the polycystic kidney disease-like channel PKD2L1
Abstract
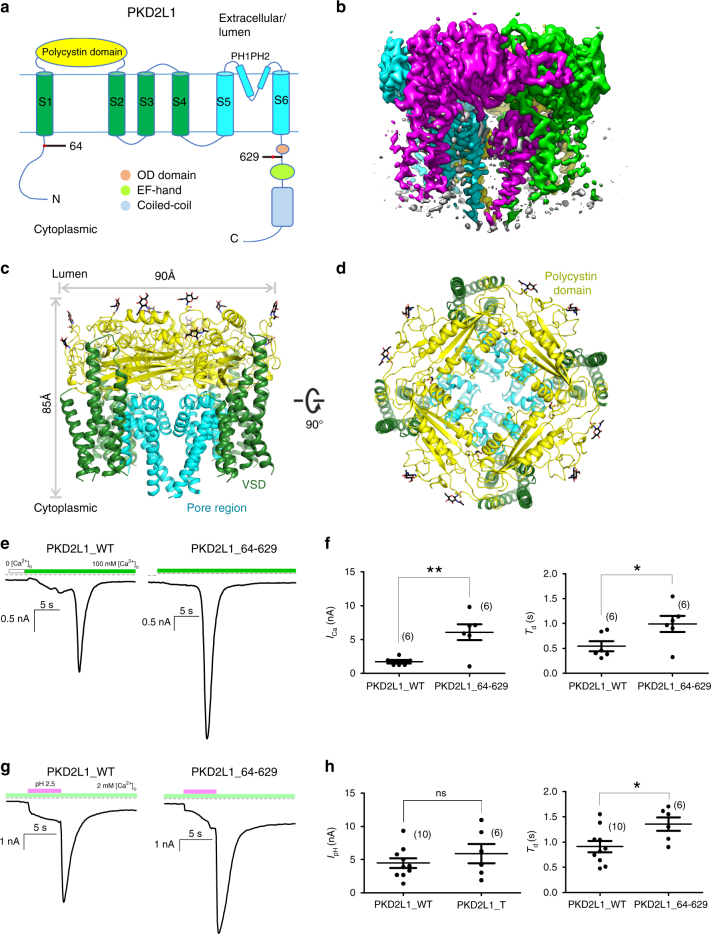
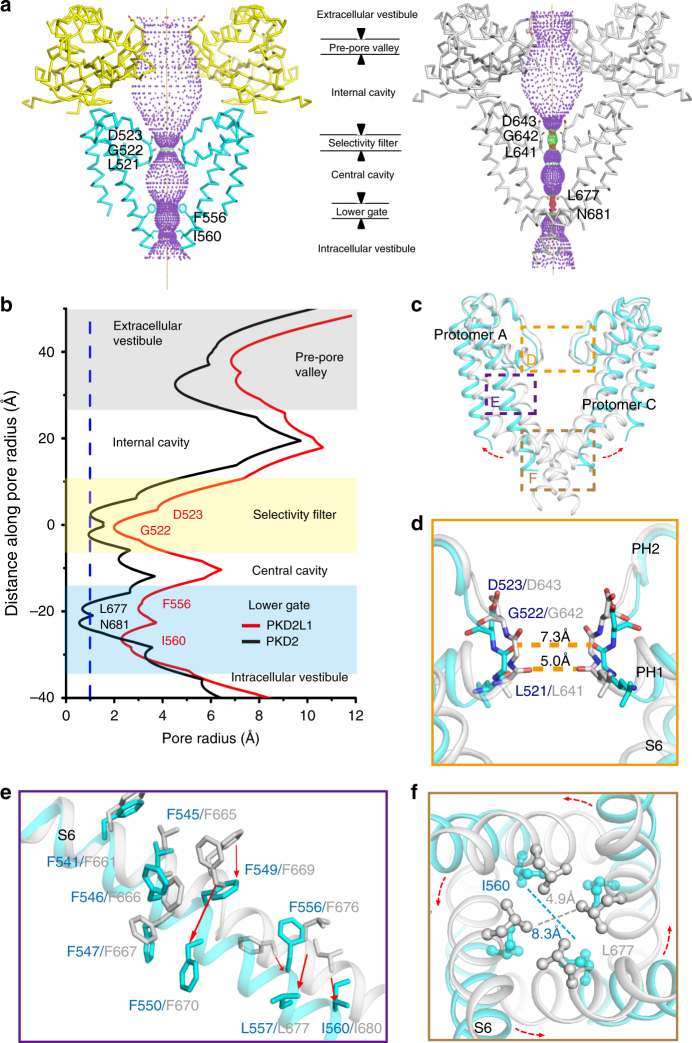
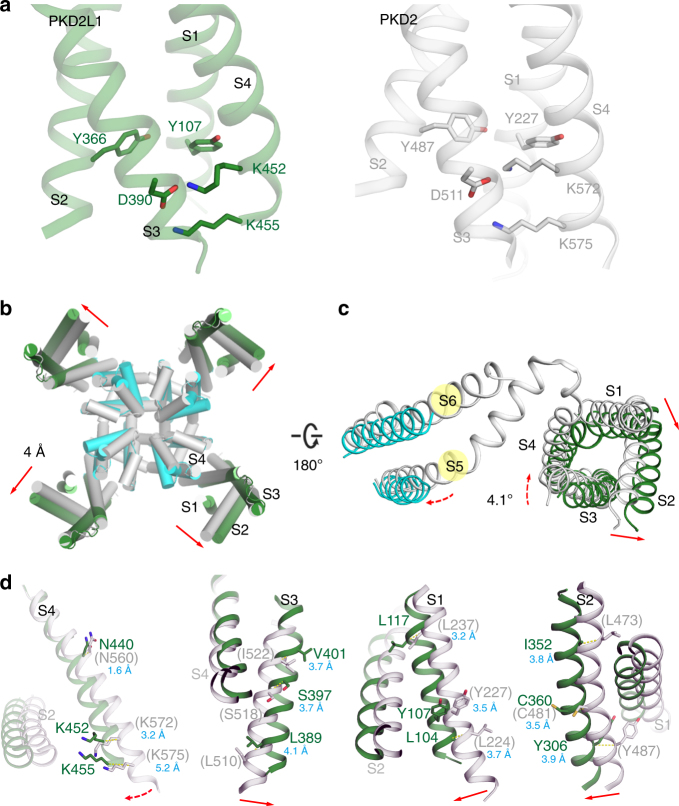
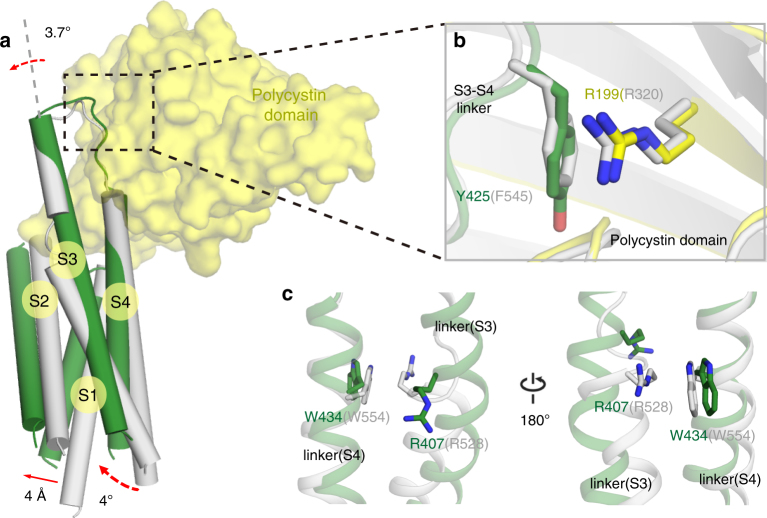
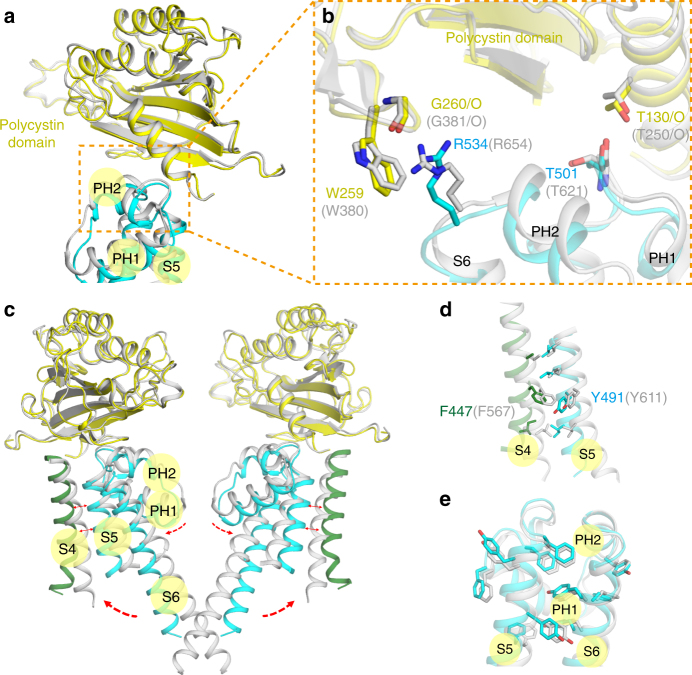
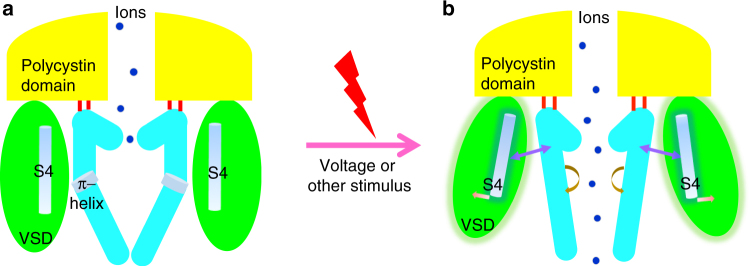
PKD2L1, also termed TRPP3 from the TRPP subfamily (polycystic TRP channels), is involved in the sour sensation and other pH-dependent processes. PKD2L1 is believed to be a nonselective cation channel that can be regulated by voltage, protons, and calcium. Despite its considerable importance, the molecular mechanisms underlying PKD2L1 regulations are largely unknown. Here, we determine the PKD2L1 atomic structure at 3.38 Å resolution by cryo-electron microscopy, whereby side chains of nearly all residues are assigned. Unlike its ortholog PKD2, the pore helix (PH) and transmembrane segment 6 (S6) of PKD2L1, which are involved in upper and lower-gate opening, adopt an open conformation. Structural comparisons of PKD2L1 with a PKD2-based homologous model indicate that the pore domain dilation is coupled to conformational changes of voltage-sensing domains (VSDs) via a series of π-π interactions, suggesting a potential PKD2L1 gating mechanism.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Structural basis for Ca2+ activation of the heteromeric PKD1L3/PKD2L1 channel.Nat Commun. 2021 Aug 11;12(1):4871. doi: 10.1038/s41467-021-25216-z. Nat Commun. 2021. PMID: 34381056 Free PMC article.
-
Hydrophobic pore gates regulate ion permeation in polycystic kidney disease 2 and 2L1 channels.Nat Commun. 2018 Jun 13;9(1):2302. doi: 10.1038/s41467-018-04586-x. Nat Commun. 2018. PMID: 29899465 Free PMC article.
-
Identification of the structural motif responsible for trimeric assembly of the C-terminal regulatory domains of polycystin channels PKD2L1 and PKD2.Biochem J. 2010 Jul 1;429(1):171-83. doi: 10.1042/BJ20091843. Biochem J. 2010. PMID: 20408813
-
Determining the Crystal Structure of TRPV6.In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. PMID: 30299652 Free Books & Documents. Review.
-
The regulatory mechanism of mammalian TRPMLs revealed by cryo-EM.FEBS J. 2018 Jul;285(14):2579-2585. doi: 10.1111/febs.14443. Epub 2018 Apr 14. FEBS J. 2018. PMID: 29577631 Free PMC article. Review.
Cited by
-
A synthetic method to assay polycystin channel biophysics.Elife. 2024 Oct 28;13:RP98534. doi: 10.7554/eLife.98534. Elife. 2024. PMID: 39466685 Free PMC article.
-
Sample Preparation for Electron Cryo-Microscopy of Macromolecular Machines.Adv Exp Med Biol. 2024;3234:173-190. doi: 10.1007/978-3-031-52193-5_12. Adv Exp Med Biol. 2024. PMID: 38507207
-
Emerging mechanistic understanding of cilia function in cellular signalling.Nat Rev Mol Cell Biol. 2024 Jul;25(7):555-573. doi: 10.1038/s41580-023-00698-5. Epub 2024 Feb 16. Nat Rev Mol Cell Biol. 2024. PMID: 38366037 Free PMC article. Review.
-
Imaging the kidney: from light to super-resolution microscopy.Nephrol Dial Transplant. 2021 Jan 1;36(1):19-28. doi: 10.1093/ndt/gfz136. Nephrol Dial Transplant. 2021. PMID: 31325314 Free PMC article. Review.
-
Lipid Interactions of a Ciliary Membrane TRP Channel: Simulation and Structural Studies of Polycystin-2.Structure. 2020 Feb 4;28(2):169-184.e5. doi: 10.1016/j.str.2019.11.005. Epub 2019 Dec 2. Structure. 2020. PMID: 31806353 Free PMC article.
References
-
- Nilius B, Flockerzi V. Mammalian transient receptor potential (TRP) cation channels. Preface. Handb. Exp. Pharmacol. 2014;223:v–vi. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
