

The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology
- PMID: 29540993
- PMCID: PMC5818912
- DOI: 10.1155/2018/1067134
The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology
Abstract
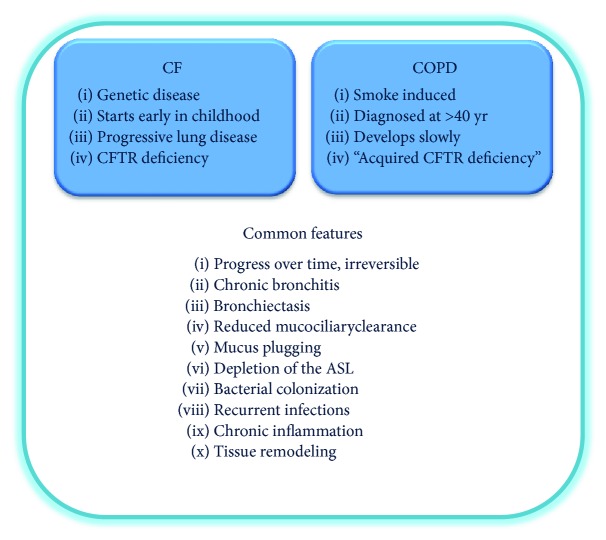
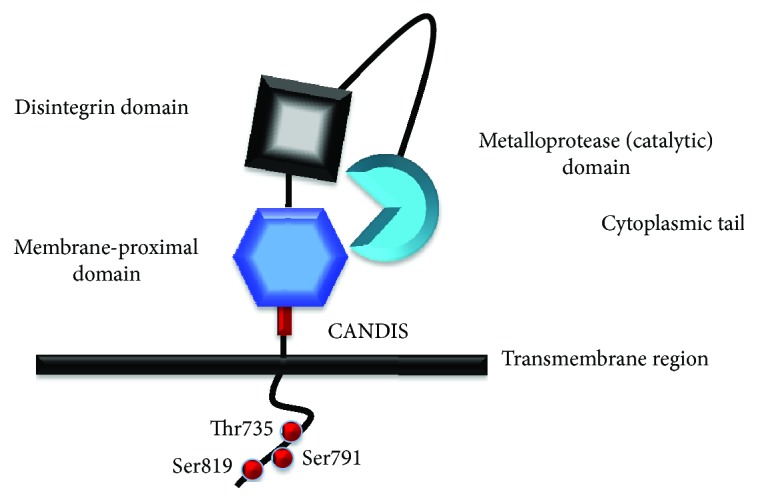
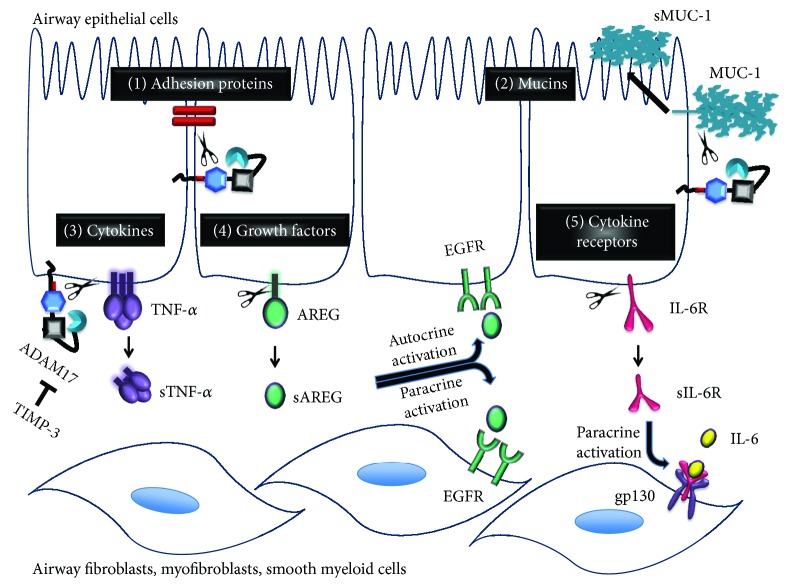
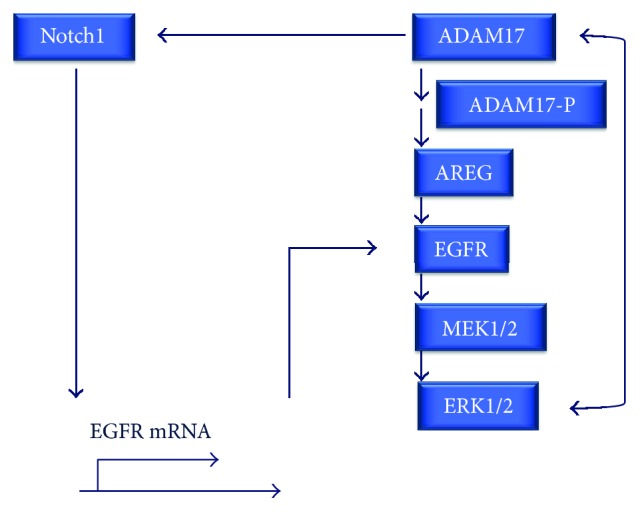
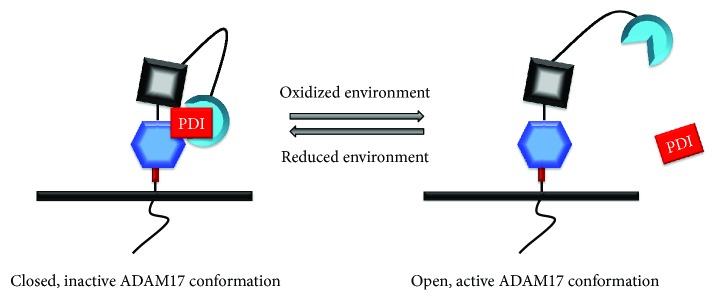
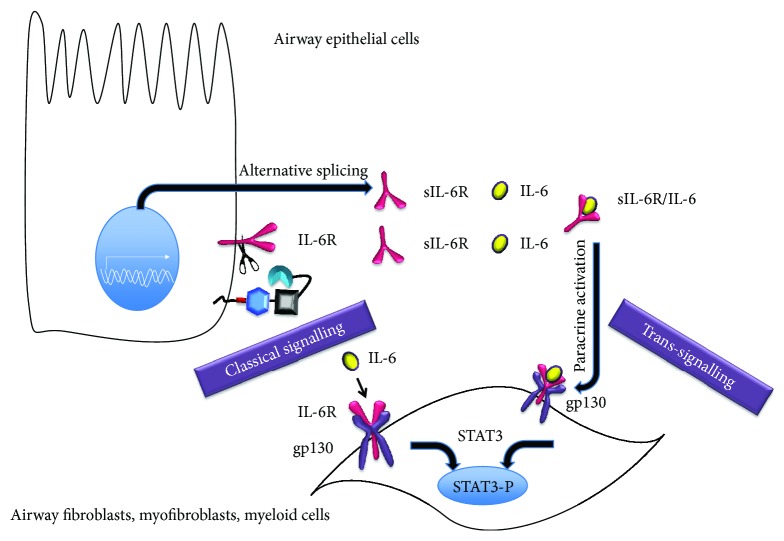
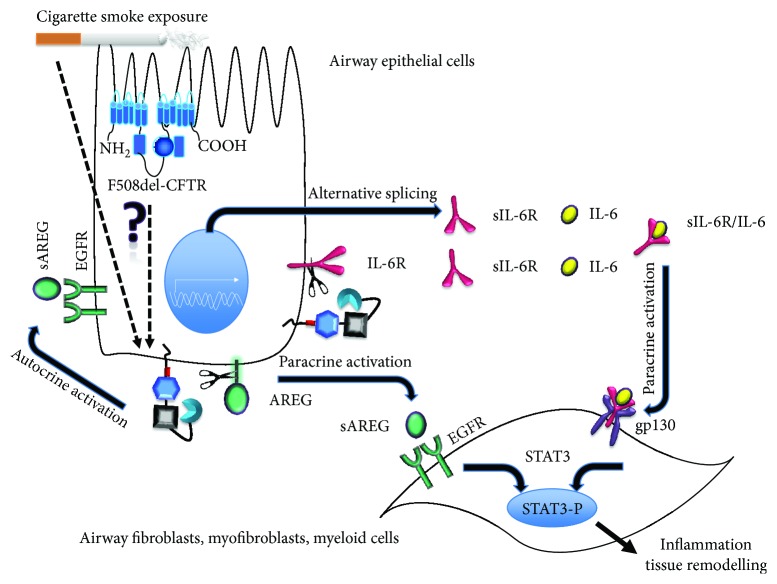
Chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF) share molecular mechanisms that cause the pathological symptoms they have in common. Here, we review evidence suggesting that hyperactivity of the EGFR/ADAM17 axis plays a role in the development of chronic lung disease in both CF and COPD. The ubiquitous transmembrane protease A disintegrin and metalloprotease 17 (ADAM17) forms a functional unit with the EGF receptor (EGFR), in a feedback loop interaction labeled the ADAM17/EGFR axis. In airway epithelial cells, ADAM17 sheds multiple soluble signaling proteins by proteolysis, including EGFR ligands such as amphiregulin (AREG), and proinflammatory mediators such as the interleukin 6 coreceptor (IL-6R). This activity can be enhanced by injury, toxins, and receptor-mediated external triggers. In addition to intracellular kinases, the extracellular glutathione-dependent redox potential controls ADAM17 shedding. Thus, the epithelial ADAM17/EGFR axis serves as a receptor of incoming luminal stress signals, relaying these to neighboring and underlying cells, which plays an important role in the resolution of lung injury and inflammation. We review evidence that congenital CFTR deficiency in CF and reduced CFTR activity in chronic COPD may cause enhanced ADAM17/EGFR signaling through a defect in glutathione secretion. In future studies, these complex interactions and the options for pharmaceutical interventions will be further investigated.
Figures
Similar articles
-
Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity.Am J Physiol Lung Cell Mol Physiol. 2018 Apr 1;314(4):L555-L568. doi: 10.1152/ajplung.00458.2017. Epub 2017 Dec 14. Am J Physiol Lung Cell Mol Physiol. 2018. PMID: 29351448
-
ADAM17 and EGFR regulate IL-6 receptor and amphiregulin mRNA expression and release in cigarette smoke-exposed primary bronchial epithelial cells from patients with chronic obstructive pulmonary disease (COPD).Physiol Rep. 2016 Aug;4(16):e12878. doi: 10.14814/phy2.12878. Physiol Rep. 2016. PMID: 27561911 Free PMC article.
-
Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin.Development. 2005 Sep;132(17):3923-33. doi: 10.1242/dev.01966. Epub 2005 Aug 3. Development. 2005. PMID: 16079154 Free PMC article.
-
The metalloprotease ADAM17 in inflammation and cancer.Pathol Res Pract. 2019 Jun;215(6):152410. doi: 10.1016/j.prp.2019.04.002. Epub 2019 Apr 6. Pathol Res Pract. 2019. PMID: 30992230 Review.
-
The role of ADAM17 during liver damage.Biol Chem. 2021 Jun 30;402(9):1115-1128. doi: 10.1515/hsz-2021-0149. Print 2021 Aug 26. Biol Chem. 2021. PMID: 34192832 Review.
Cited by
-
Alpha-1-antitrypsin: A possible host protective factor against Covid-19.Rev Med Virol. 2021 Mar;31(2):e2157. doi: 10.1002/rmv.2157. Epub 2020 Aug 26. Rev Med Virol. 2021. PMID: 32844538 Free PMC article. Review.
-
The Downregulation of ADAM17 Exerts Protective Effects against Cardiac Fibrosis by Regulating Endoplasmic Reticulum Stress and Mitophagy.Oxid Med Cell Longev. 2021 May 6;2021:5572088. doi: 10.1155/2021/5572088. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34035876 Free PMC article.
-
Immunomodulatory role of metalloproteinase ADAM17 in tumor development.Front Immunol. 2022 Nov 17;13:1059376. doi: 10.3389/fimmu.2022.1059376. eCollection 2022. Front Immunol. 2022. PMID: 36466812 Free PMC article. Review.
-
Gefitinib and fostamatinib target EGFR and SYK to attenuate silicosis: a multi-omics study with drug exploration.Signal Transduct Target Ther. 2022 May 13;7(1):157. doi: 10.1038/s41392-022-00959-3. Signal Transduct Target Ther. 2022. PMID: 35551173 Free PMC article.
-
Mechanisms of Virus-Induced Airway Immunity Dysfunction in the Pathogenesis of COPD Disease, Progression, and Exacerbation.Front Immunol. 2020 Jun 16;11:1205. doi: 10.3389/fimmu.2020.01205. eCollection 2020. Front Immunol. 2020. PMID: 32655557 Free PMC article. Review.
References
-
- Gomez M. I., Sokol S. H., Muir A. B., Soong G., Bastien J., Prince A. S. Bacterial induction of TNF-alpha converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. The Journal of Immunology. 2005;175(3):1930–1936. doi: 10.4049/jimmunol.175.3.1930. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
