

Endoplasmic Reticulum Protein TXNDC5 Augments Myocardial Fibrosis by Facilitating Extracellular Matrix Protein Folding and Redox-Sensitive Cardiac Fibroblast Activation
- PMID: 29535165
- PMCID: PMC5899016
- DOI: 10.1161/CIRCRESAHA.117.312130
Endoplasmic Reticulum Protein TXNDC5 Augments Myocardial Fibrosis by Facilitating Extracellular Matrix Protein Folding and Redox-Sensitive Cardiac Fibroblast Activation
Abstract
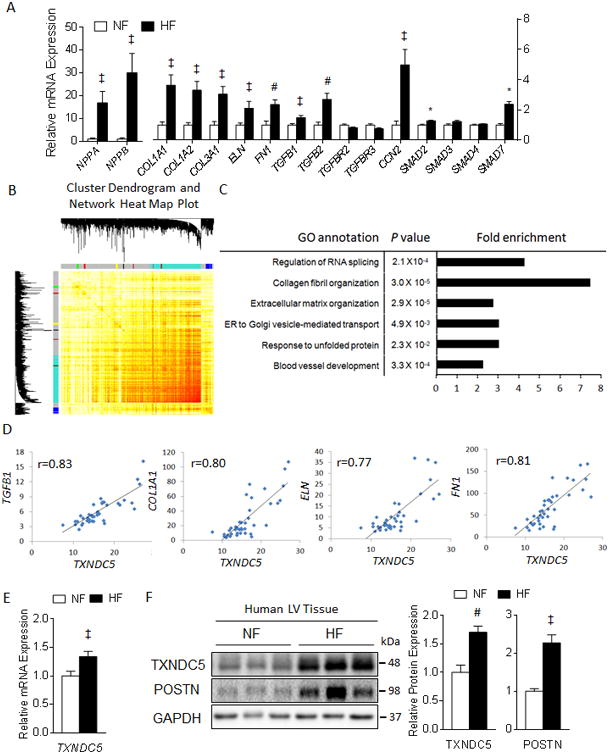
Rationale: Cardiac fibrosis plays a critical role in the pathogenesis of heart failure. Excessive accumulation of extracellular matrix (ECM) resulting from cardiac fibrosis impairs cardiac contractile function and increases arrhythmogenicity. Current treatment options for cardiac fibrosis, however, are limited, and there is a clear need to identify novel mediators of cardiac fibrosis to facilitate the development of better therapeutics. Exploiting coexpression gene network analysis on RNA sequencing data from failing human heart, we identified TXNDC5 (thioredoxin domain containing 5), a cardiac fibroblast (CF)-enriched endoplasmic reticulum protein, as a potential novel mediator of cardiac fibrosis, and we completed experiments to test this hypothesis directly.
Objective: The objective of this study was to determine the functional role of TXNDC5 in the pathogenesis of cardiac fibrosis.
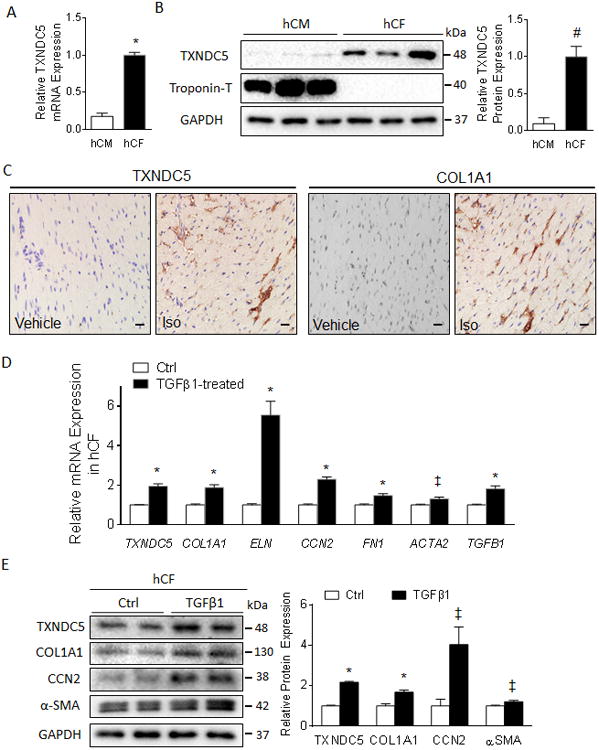
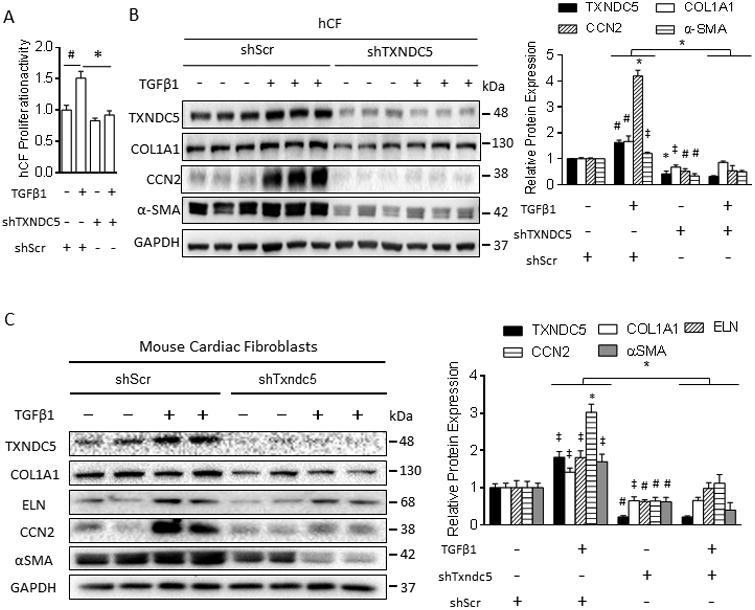
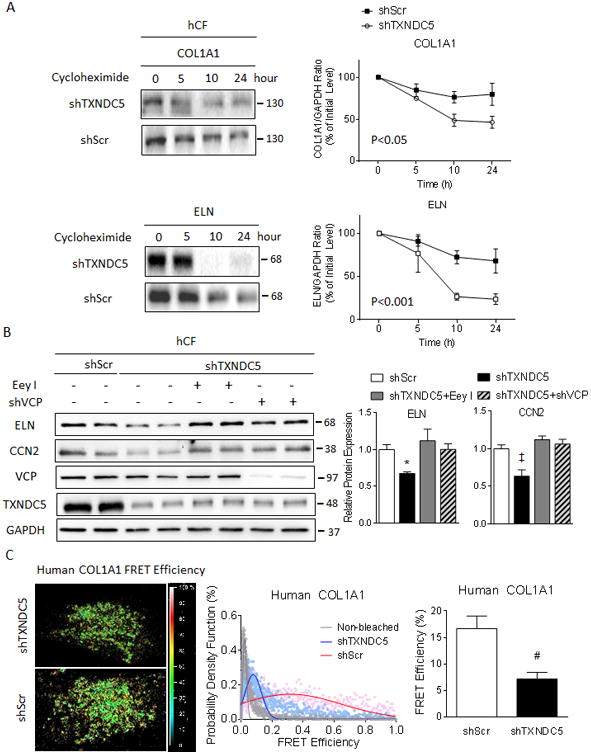
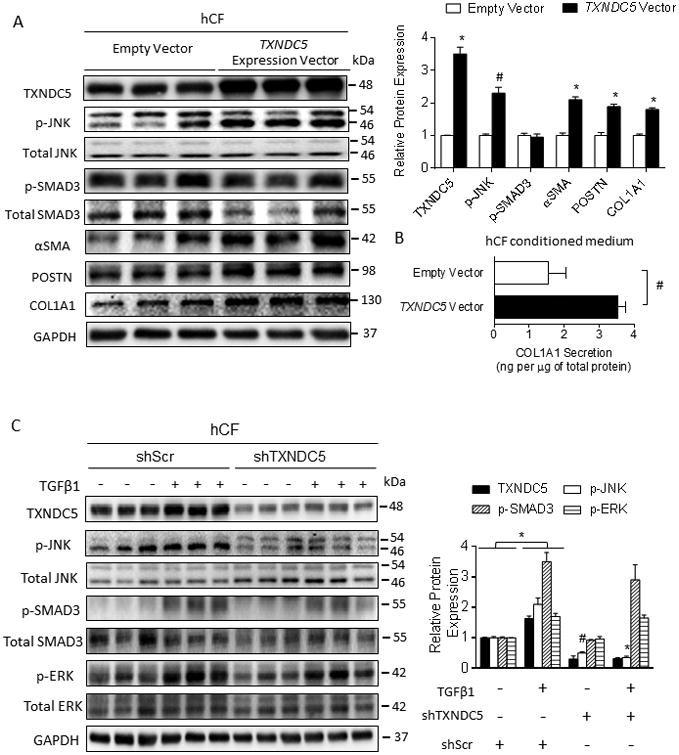
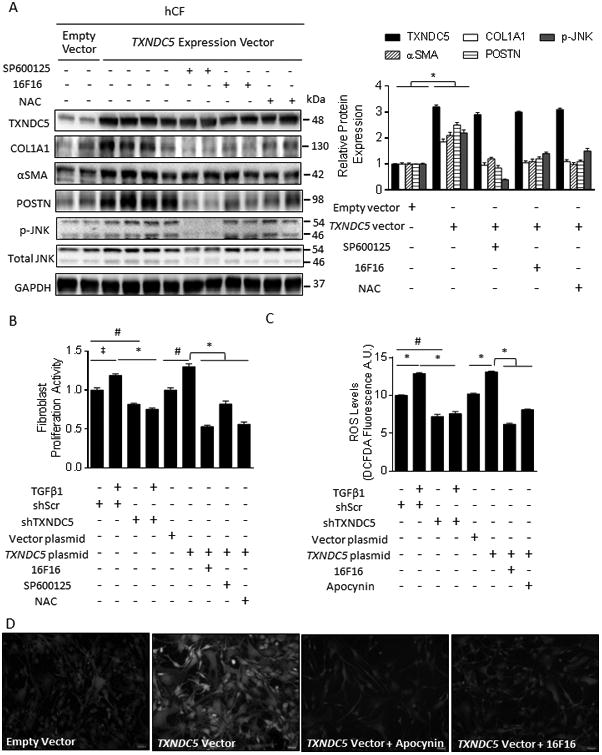
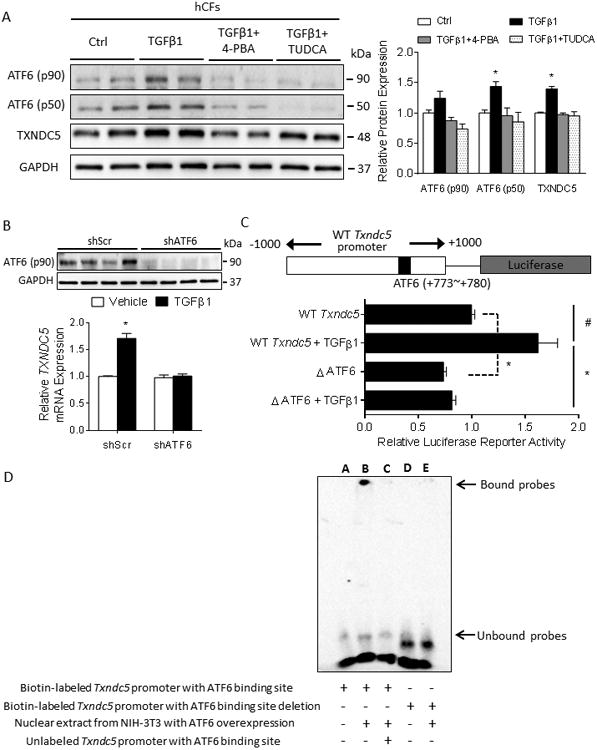
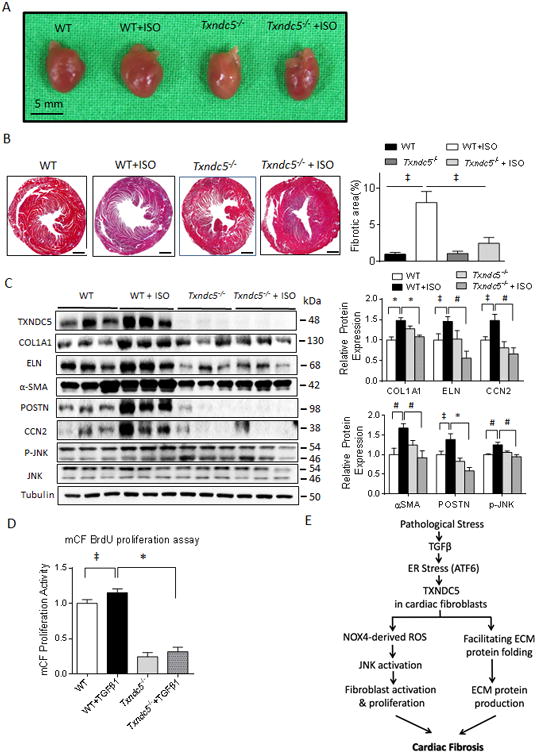
Methods and results: RNA sequencing and Western blot analyses revealed that TXNDC5 mRNA and protein were highly upregulated in failing human left ventricles and in hypertrophied/failing mouse left ventricle. In addition, cardiac TXNDC5 mRNA expression levels were positively correlated with those of transcripts encoding transforming growth factor β1 and ECM proteins in vivo. TXNDC5 mRNA and protein were increased in human CF (hCF) under transforming growth factor β1 stimulation in vitro. Knockdown of TXNDC5 attenuated transforming growth factor β1-induced hCF activation and ECM protein upregulation independent of SMAD3 (SMAD family member 3), whereas increasing expression of TXNDC5 triggered hCF activation and proliferation and increased ECM protein production. Further experiments showed that TXNDC5, a protein disulfide isomerase, facilitated ECM protein folding and that depletion of TXNDC5 led to ECM protein misfolding and degradation in CF. In addition, TXNDC5 promotes hCF activation and proliferation by enhancing c-Jun N-terminal kinase activity via increased reactive oxygen species, derived from NAD(P)H oxidase 4. Transforming growth factor β1-induced TXNDC5 upregulation in hCF was dependent on endoplasmic reticulum stress and activating transcription factor 6-mediated transcriptional control. Targeted disruption of Txndc5 in mice (Txndc5-/-) revealed protective effects against isoproterenol-induced cardiac hypertrophy, reduced fibrosis (by ≈70%), and markedly improved left ventricle function; post-isoproterenol left ventricular ejection fraction was 59.1±1.5 versus 40.1±2.5 (P<0.001) in Txndc5-/- versus wild-type mice, respectively.
Conclusions: The endoplasmic reticulum protein TXNDC5 promotes cardiac fibrosis by facilitating ECM protein folding and CF activation via redox-sensitive c-Jun N-terminal kinase signaling. Loss of TXNDC5 protects against β agonist-induced cardiac fibrosis and contractile dysfunction. Targeting TXNDC5, therefore, could be a powerful new therapeutic approach to mitigate excessive cardiac fibrosis, thereby improving cardiac function and outcomes in patients with heart failure.
Keywords: endoplasmic reticulum; fibrosis; heart failure; oxidative stress; sequence analysis, RNA.
© 2018 American Heart Association, Inc.
Figures
Similar articles
-
Fibroblast-enriched endoplasmic reticulum protein TXNDC5 promotes pulmonary fibrosis by augmenting TGFβ signaling through TGFBR1 stabilization.Nat Commun. 2020 Aug 26;11(1):4254. doi: 10.1038/s41467-020-18047-x. Nat Commun. 2020. PMID: 32848143 Free PMC article.
-
Endoplasmic reticulum protein TXNDC5 promotes renal fibrosis by enforcing TGF-β signaling in kidney fibroblasts.J Clin Invest. 2021 Mar 1;131(5):e143645. doi: 10.1172/JCI143645. J Clin Invest. 2021. PMID: 33465051 Free PMC article.
-
Endoplasmic reticulum protein TXNDC5 modulates thyroid eye disease TGF-β1-induced myofibroblast transdifferentiation.BMJ Open Ophthalmol. 2024 Dec 25;9(1):e001693. doi: 10.1136/bmjophth-2024-001693. BMJ Open Ophthalmol. 2024. PMID: 39721966 Free PMC article.
-
The novel role of ER protein TXNDC5 in the pathogenesis of organ fibrosis: mechanistic insights and therapeutic implications.J Biomed Sci. 2022 Sep 2;29(1):63. doi: 10.1186/s12929-022-00850-x. J Biomed Sci. 2022. PMID: 36050716 Free PMC article. Review.
-
Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis.Cardiovasc Res. 2000 May;46(2):257-63. doi: 10.1016/s0008-6363(00)00030-4. Cardiovasc Res. 2000. PMID: 10773229 Review.
Cited by
-
Intermedin1-53 Inhibits NLRP3 Inflammasome Activation by Targeting IRE1α in Cardiac Fibrosis.Inflammation. 2022 Aug;45(4):1568-1584. doi: 10.1007/s10753-022-01642-z. Epub 2022 Feb 17. Inflammation. 2022. PMID: 35175495
-
Integrative analysis of genes reveals endoplasmic reticulum stress-related immune responses involved in dilated cardiomyopathy with fibrosis.Apoptosis. 2023 Oct;28(9-10):1406-1421. doi: 10.1007/s10495-023-01871-z. Epub 2023 Jul 18. Apoptosis. 2023. PMID: 37462883 Free PMC article.
-
Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved.Cells. 2021 Jul 19;10(7):1824. doi: 10.3390/cells10071824. Cells. 2021. PMID: 34359993 Free PMC article. Review.
-
A clear pathway to tubulointerstitial disease: is an exclusive focus on fibrosis justified?J Clin Invest. 2021 Mar 1;131(5):e144803. doi: 10.1172/JCI144803. J Clin Invest. 2021. PMID: 33645547 Free PMC article.
-
Thioredoxin Domain Containing 5 Suppression Elicits Serum Amyloid A-Containing High-Density Lipoproteins.Biomedicines. 2022 Mar 18;10(3):709. doi: 10.3390/biomedicines10030709. Biomedicines. 2022. PMID: 35327511 Free PMC article.
References
-
- McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J. 1998;19(P):P9–16. - PubMed
-
- Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. - PubMed
-
- Braunwald E. Shattuck lecture--cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med. 1997;337:1360–9. - PubMed
-
- Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–402. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
