

Innate immune responses to trauma
- PMID: 29507356
- PMCID: PMC6027646
- DOI: 10.1038/s41590-018-0064-8
Innate immune responses to trauma
Abstract
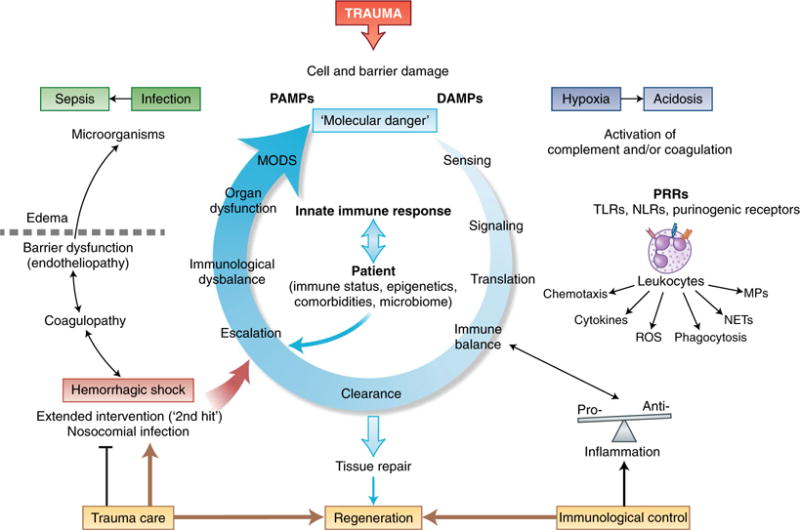
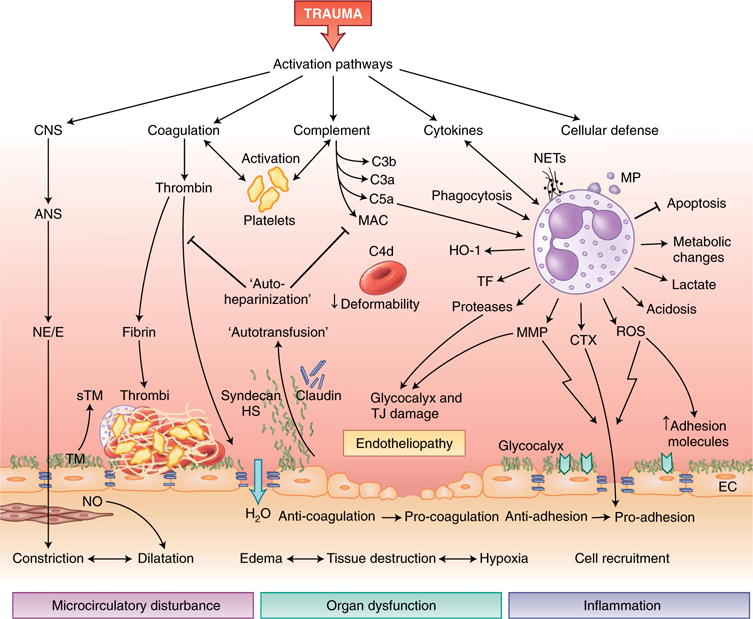
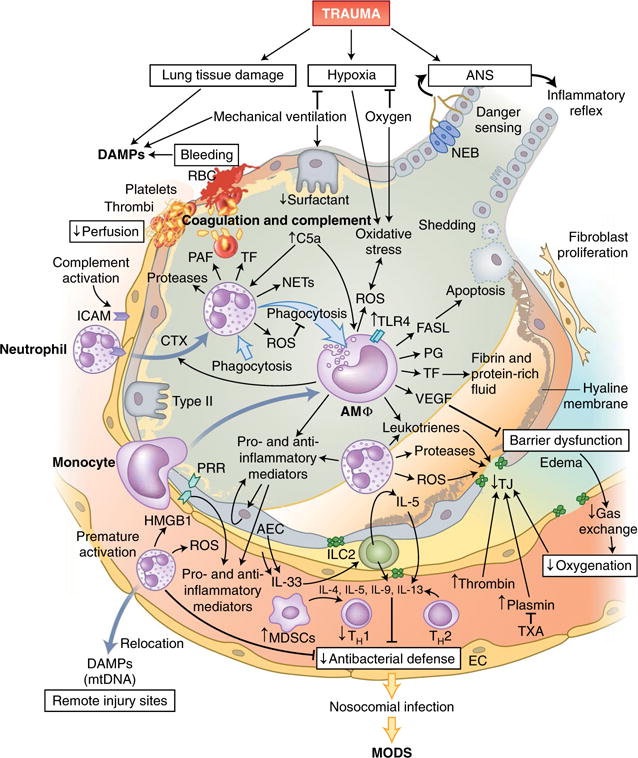
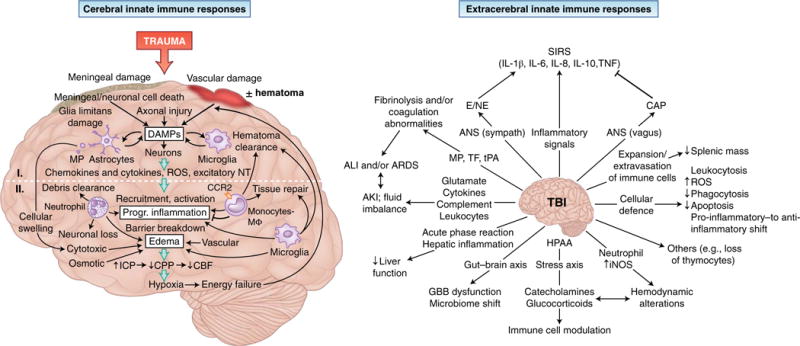
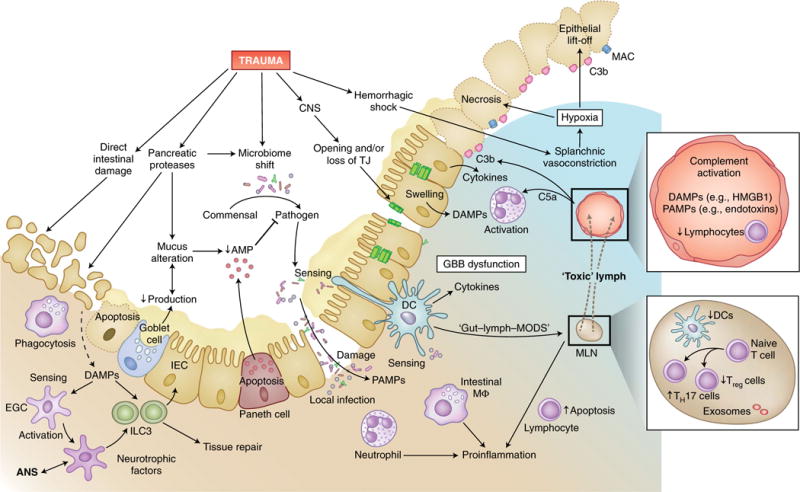
Trauma can affect any individual at any location and at any time over a lifespan. The disruption of macrobarriers and microbarriers induces instant activation of innate immunity. The subsequent complex response, designed to limit further damage and induce healing, also represents a major driver of complications and fatal outcome after injury. This Review aims to provide basic concepts about the posttraumatic response and is focused on the interactive events of innate immunity at frequent sites of injury: the endothelium at large, and sites within the lungs, inside and outside the brain and at the gut barrier.
Conflict of interest statement
M.H.-L. and P.A.W. hold a patent on compositions and methods for the diagnosis and treatment of sepsis (US 7455837). J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs such as AMY-101), and is the inventor of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. J.D.L. is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives such as APL-2).
Figures
Similar articles
-
Innate immune dysfunction in trauma patients: from pathophysiology to treatment.Anesthesiology. 2012 Aug;117(2):411-6. doi: 10.1097/ALN.0b013e31825f018d. Anesthesiology. 2012. PMID: 22728780 Review. No abstract available.
-
Complement After Trauma: Suturing Innate and Adaptive Immunity.Front Immunol. 2018 Sep 24;9:2050. doi: 10.3389/fimmu.2018.02050. eCollection 2018. Front Immunol. 2018. PMID: 30319602 Free PMC article. Review.
-
Molecular mechanisms of inflammation and tissue injury after major trauma--is complement the "bad guy"?J Biomed Sci. 2011 Nov 30;18(1):90. doi: 10.1186/1423-0127-18-90. J Biomed Sci. 2011. PMID: 22129197 Free PMC article. Review.
-
IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing.J Invest Dermatol. 2016 Feb;136(2):487-496. doi: 10.1038/JID.2015.406. J Invest Dermatol. 2016. PMID: 26802241 Free PMC article.
-
Androstenediol exerts salutary effects on chemokine response after trauma-hemorrhage and sepsis in mice.J Orthop Trauma. 2011 Aug;25(8):511-5. doi: 10.1097/BOT.0b013e3182251044. J Orthop Trauma. 2011. PMID: 21738064
Cited by
-
Plasma interleukin responses as predictors of outcome stratification in patients after major trauma: a prospective observational two centre study.Front Immunol. 2023 Nov 23;14:1276171. doi: 10.3389/fimmu.2023.1276171. eCollection 2023. Front Immunol. 2023. PMID: 38077362 Free PMC article.
-
Adipose tissue: a neglected organ in the response to severe trauma?Cell Mol Life Sci. 2022 Mar 26;79(4):207. doi: 10.1007/s00018-022-04234-0. Cell Mol Life Sci. 2022. PMID: 35338424 Free PMC article. Review.
-
Changes in the microbiota in different intestinal segments of mice with sepsis.Front Cell Infect Microbiol. 2023 Jan 10;12:954347. doi: 10.3389/fcimb.2022.954347. eCollection 2022. Front Cell Infect Microbiol. 2023. PMID: 36704101 Free PMC article.
-
Crosstalk Between Autophagy and Inflammation in Chronic Cerebral Ischaemia.Cell Mol Neurobiol. 2023 Aug;43(6):2557-2566. doi: 10.1007/s10571-023-01336-6. Epub 2023 Mar 23. Cell Mol Neurobiol. 2023. PMID: 36952071 Free PMC article. Review.
-
The Role of microRNA in the Inflammatory Response of Wound Healing.Front Immunol. 2022 Mar 21;13:852419. doi: 10.3389/fimmu.2022.852419. eCollection 2022. Front Immunol. 2022. PMID: 35386721 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
