

Metabolic Reprogramming in Amyotrophic Lateral Sclerosis
- PMID: 29500423
- PMCID: PMC5834494
- DOI: 10.1038/s41598-018-22318-5
Metabolic Reprogramming in Amyotrophic Lateral Sclerosis
Abstract
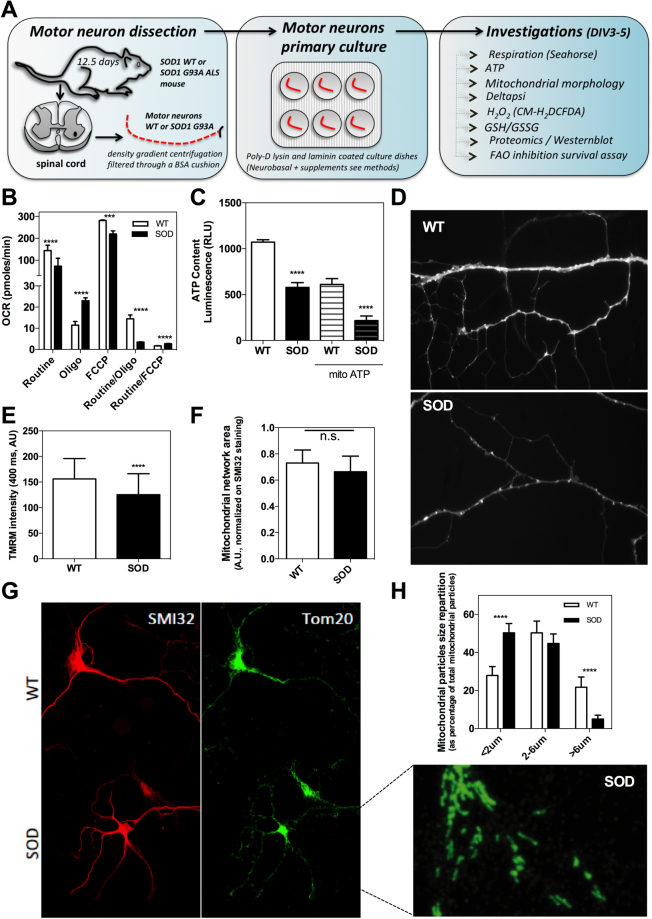
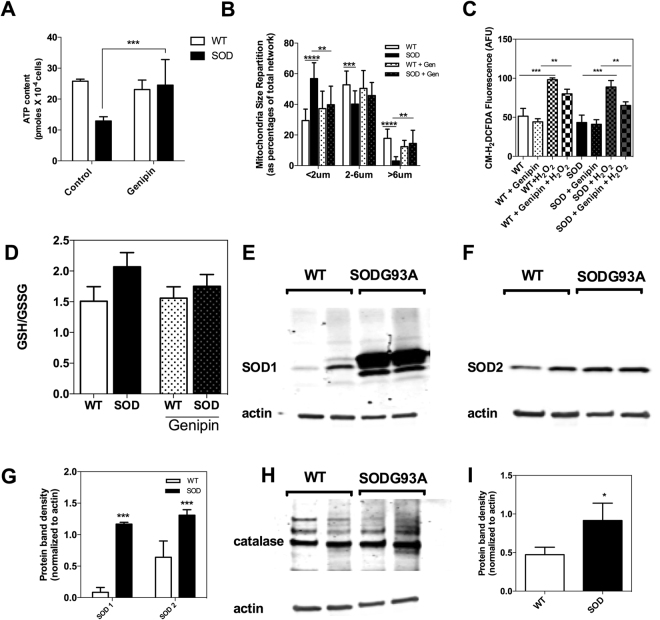
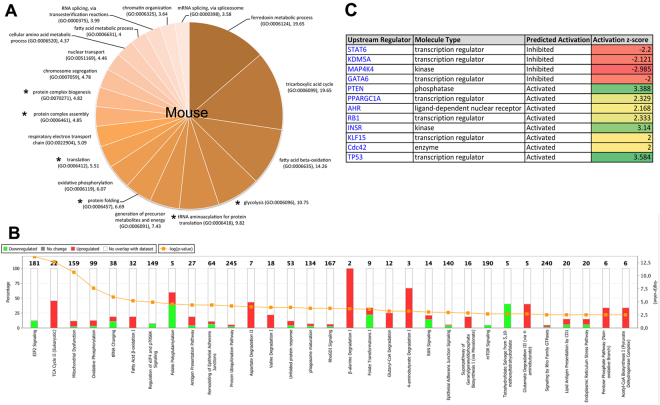
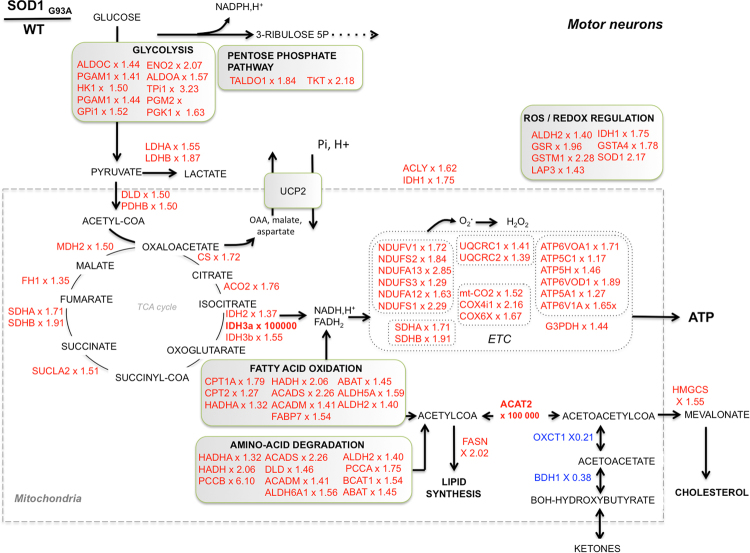
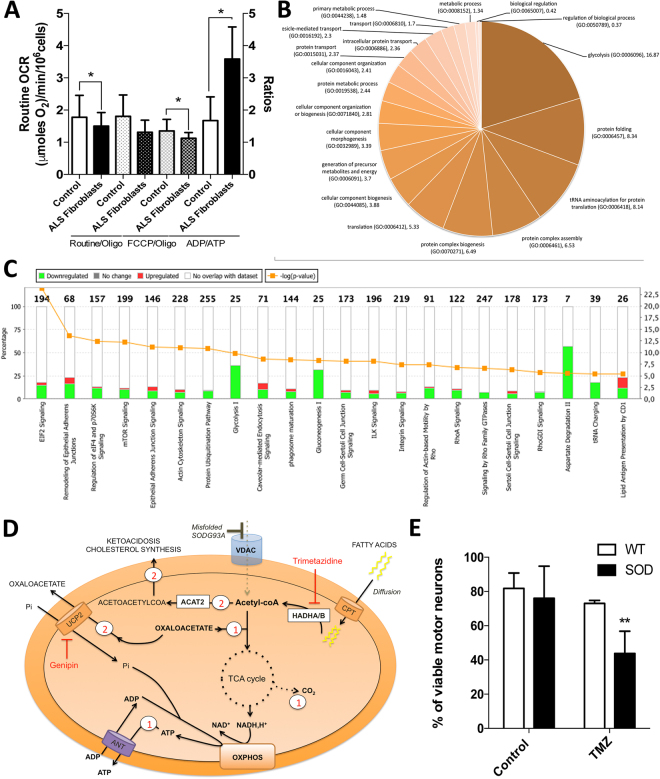
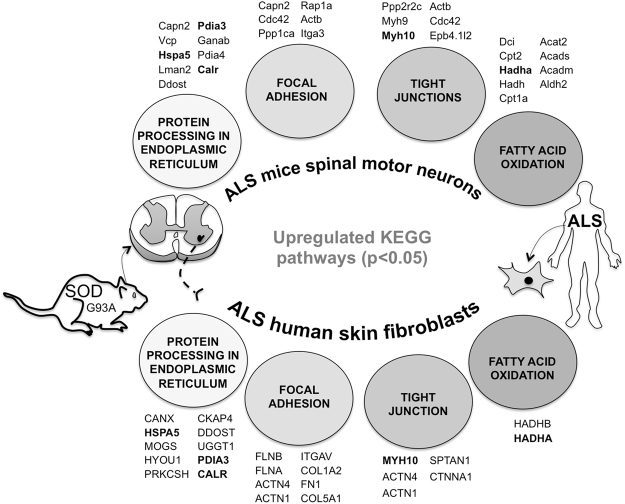
Mitochondrial dysfunction in the spinal cord is a hallmark of amyotrophic lateral sclerosis (ALS), but the neurometabolic alterations during early stages of the disease remain unknown. Here, we investigated the bioenergetic and proteomic changes in ALS mouse motor neurons and patients' skin fibroblasts. We first observed that SODG93A mice presymptomatic motor neurons display alterations in the coupling efficiency of oxidative phosphorylation, along with fragmentation of the mitochondrial network. The proteome of presymptomatic ALS mice motor neurons also revealed a peculiar metabolic signature with upregulation of most energy-transducing enzymes, including the fatty acid oxidation (FAO) and the ketogenic components HADHA and ACAT2, respectively. Accordingly, FAO inhibition altered cell viability specifically in ALS mice motor neurons, while uncoupling protein 2 (UCP2) inhibition recovered cellular ATP levels and mitochondrial network morphology. These findings suggest a novel hypothesis of ALS bioenergetics linking FAO and UCP2. Lastly, we provide a unique set of data comparing the molecular alterations found in human ALS patients' skin fibroblasts and SODG93A mouse motor neurons, revealing conserved changes in protein translation, folding and assembly, tRNA aminoacylation and cell adhesion processes.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis.Neurobiol Aging. 2014 Jun;35(6):1499-509. doi: 10.1016/j.neurobiolaging.2013.11.025. Epub 2013 Dec 3. Neurobiol Aging. 2014. PMID: 24439480
-
Comparing effects of microgravity and amyotrophic lateral sclerosis in the mouse ventral lumbar spinal cord.Mol Cell Neurosci. 2022 Jul;121:103745. doi: 10.1016/j.mcn.2022.103745. Epub 2022 Jun 2. Mol Cell Neurosci. 2022. PMID: 35660087
-
Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis.Hum Mol Genet. 2009 Jan 1;18(1):82-96. doi: 10.1093/hmg/ddn319. Epub 2008 Sep 29. Hum Mol Genet. 2009. PMID: 18826962 Free PMC article.
-
AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis.Neurochem Res. 2016 Mar;41(3):544-53. doi: 10.1007/s11064-015-1665-3. Epub 2015 Jul 23. Neurochem Res. 2016. PMID: 26202426 Review.
-
Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target?Mol Neurobiol. 2006 Apr;33(2):113-31. doi: 10.1385/MN:33:2:113. Mol Neurobiol. 2006. PMID: 16603792 Review.
Cited by
-
Diet, Microbiota and Brain Health: Unraveling the Network Intersecting Metabolism and Neurodegeneration.Int J Mol Sci. 2020 Oct 10;21(20):7471. doi: 10.3390/ijms21207471. Int J Mol Sci. 2020. PMID: 33050475 Free PMC article. Review.
-
Gut microbiome correlates with plasma lipids in amyotrophic lateral sclerosis.Brain. 2024 Feb 1;147(2):665-679. doi: 10.1093/brain/awad306. Brain. 2024. PMID: 37721161 Free PMC article.
-
Amyotrophic lateral sclerosis alters the metabolic aging profile in patient derived fibroblasts.Neurobiol Aging. 2021 Sep;105:64-77. doi: 10.1016/j.neurobiolaging.2021.04.013. Epub 2021 Apr 27. Neurobiol Aging. 2021. PMID: 34044197 Free PMC article.
-
The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders.Transl Neurodegener. 2022 Jul 4;11(1):36. doi: 10.1186/s40035-022-00308-y. Transl Neurodegener. 2022. PMID: 35787292 Free PMC article. Review.
-
The Relationship between Body Composition, Fatty Acid Metabolism and Diet in Spinal Muscular Atrophy.Brain Sci. 2021 Jan 20;11(2):131. doi: 10.3390/brainsci11020131. Brain Sci. 2021. PMID: 33498293 Free PMC article. Review.
References
-
- Karbowski, M. & Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol, 10.1007/s00401-011-0921-0 (2011). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
