

Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation
- PMID: 29462784
- PMCID: PMC5892583
- DOI: 10.1074/jbc.M117.811075
Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation
Abstract
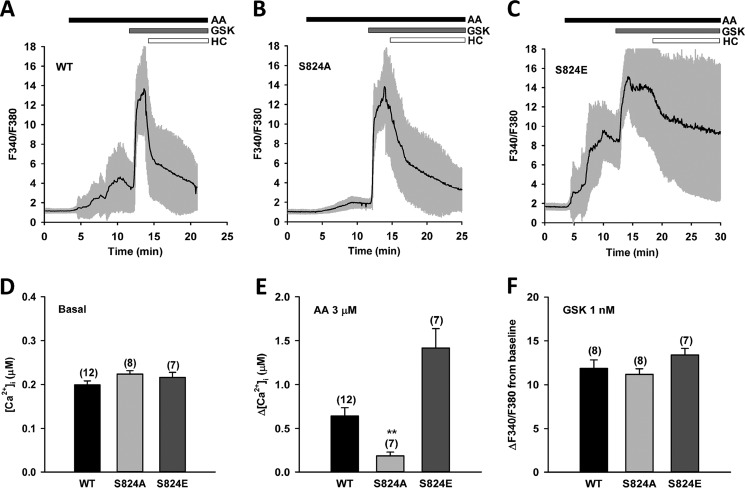
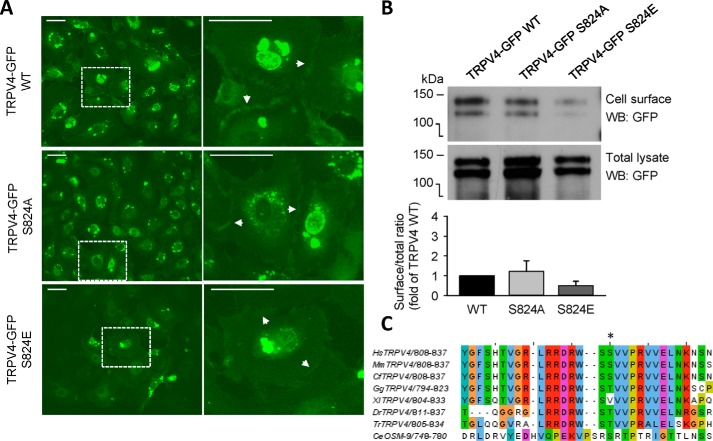
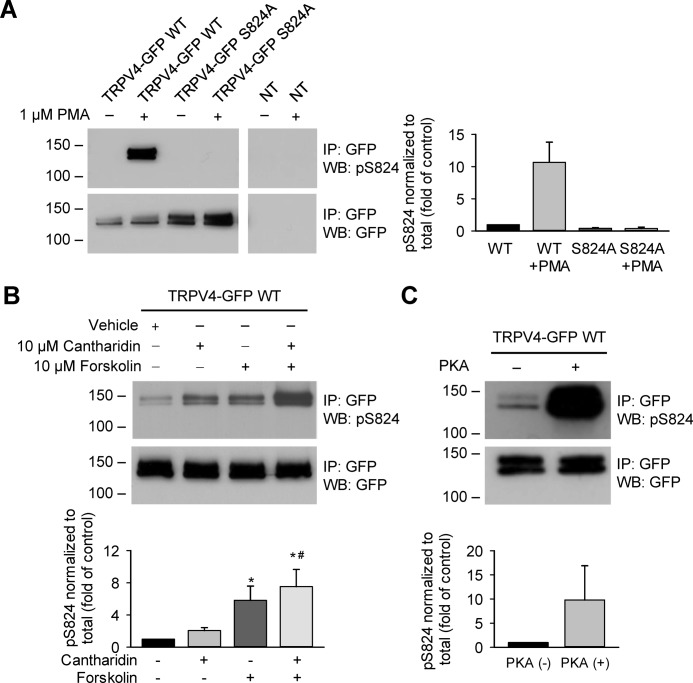
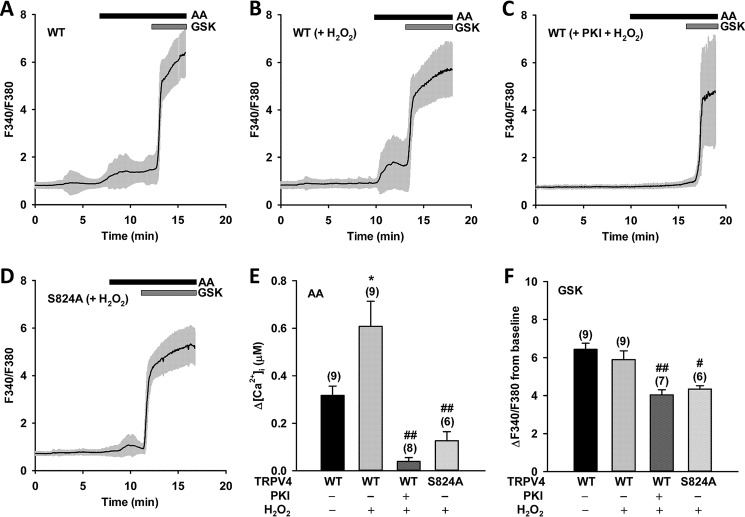
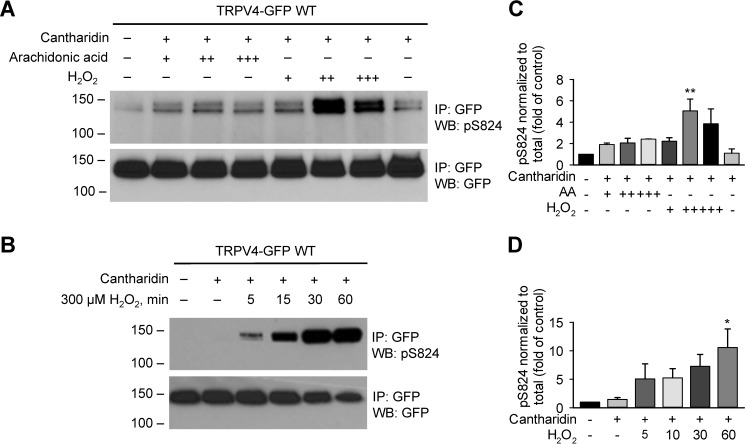
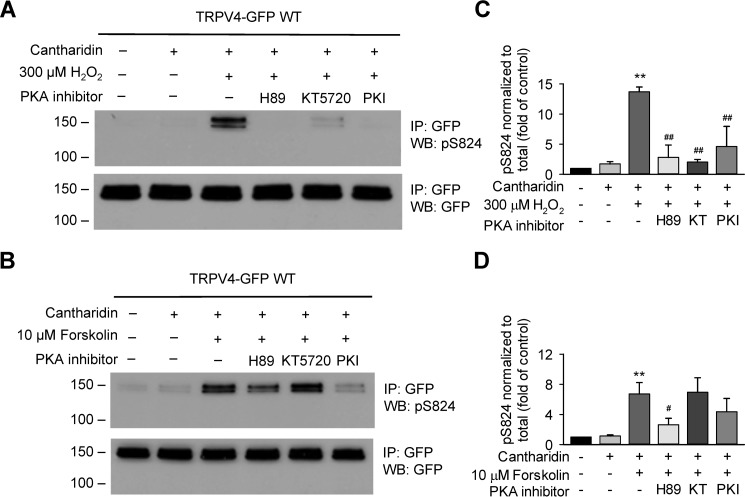
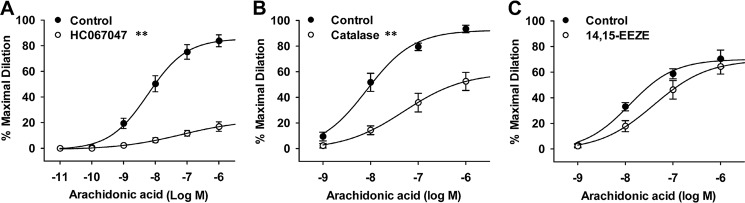
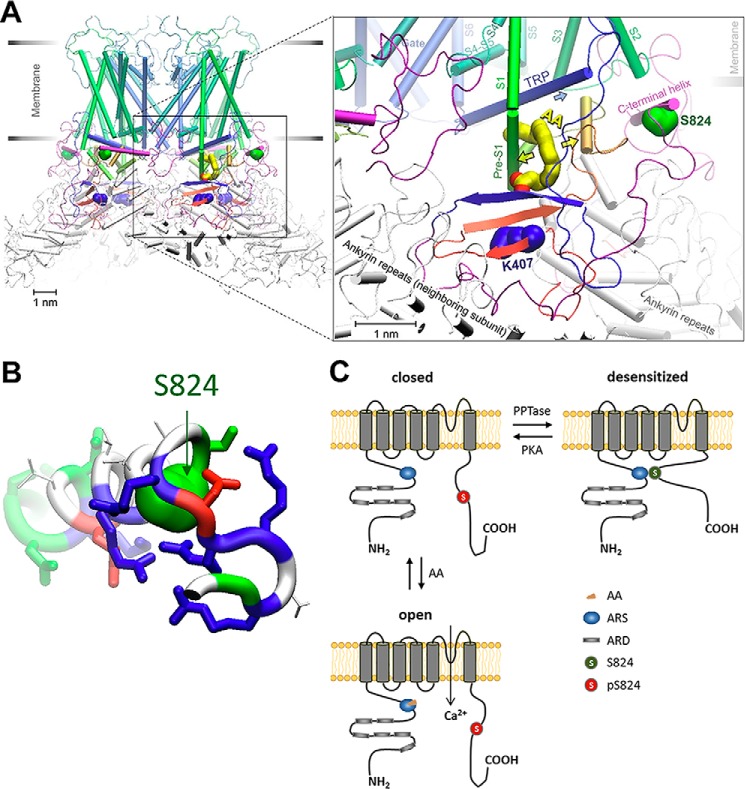
Transient receptor potential vanilloid 4 (TRPV4) is a Ca2+-permeable channel of the transient receptor potential (TRP) superfamily activated by diverse stimuli, including warm temperature, mechanical forces, and lipid mediators such as arachidonic acid (AA) and its metabolites. This activation is tightly regulated by protein phosphorylation carried out by various serine/threonine or tyrosine kinases. It remains poorly understood how phosphorylation differentially regulates TRPV4 activation in response to different stimuli. We investigated how TRPV4 activation by AA, an important signaling process in the dilation of coronary arterioles, is affected by protein kinase A (PKA)-mediated phosphorylation at Ser-824. Wildtype and mutant TRPV4 channels were expressed in human coronary artery endothelial cells (HCAECs). AA-induced TRPV4 activation was blunted in the S824A mutant but was enhanced in the phosphomimetic S824E mutant, whereas the channel activation by the synthetic agonist GSK1016790A was not affected. The low level of basal phosphorylation at Ser-824 was robustly increased by the redox signaling molecule hydrogen peroxide (H2O2). The H2O2-induced phosphorylation was accompanied by an enhanced channel activation by AA, and this enhanced response was largely abolished by PKA inhibition or S824A mutation. We further identified a potential structural context dependence of Ser-824 phosphorylation-mediated TRPV4 regulation involving an interplay between AA binding and the possible phosphorylation-induced rearrangements of the C-terminal helix bearing Ser-824. These results provide insight into how phosphorylation specifically regulates TRPV4 activation. Redox-mediated TRPV4 phosphorylation may contribute to pathologies associated with enhanced TRPV4 activity in endothelial and other systems.
Keywords: arachidonic acid (AA) (ARA); endothelial cell; hydrogen peroxide; protein kinase A (PKA); protein phosphorylation; signal transduction; transient receptor potential channels (TRP channels).
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry.J Am Heart Assoc. 2013 Apr 25;2(3):e000080. doi: 10.1161/JAHA.113.000080. J Am Heart Assoc. 2013. PMID: 23619744 Free PMC article.
-
Activation of the TRPV4 ion channel is enhanced by phosphorylation.J Biol Chem. 2009 Oct 9;284(41):27884-27891. doi: 10.1074/jbc.M109.028803. Epub 2009 Aug 6. J Biol Chem. 2009. PMID: 19661060 Free PMC article.
-
Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling.Am J Physiol Heart Circ Physiol. 2012 Feb 1;302(3):H634-42. doi: 10.1152/ajpheart.00717.2011. Epub 2011 Dec 2. Am J Physiol Heart Circ Physiol. 2012. PMID: 22140047 Free PMC article.
-
Transient receptor potential vanilloid 4: The sixth sense of the musculoskeletal system?Ann N Y Acad Sci. 2010 Mar;1192:404-9. doi: 10.1111/j.1749-6632.2010.05389.x. Ann N Y Acad Sci. 2010. PMID: 20392266 Free PMC article. Review.
-
TRP channels activated by extracellular hypo-osmoticity in epithelia.Biochem Soc Trans. 2007 Feb;35(Pt 1):91-5. doi: 10.1042/BST0350091. Biochem Soc Trans. 2007. PMID: 17233610 Review.
Cited by
-
TRPV4 Channel Modulators as Potential Drug Candidates for Cystic Fibrosis.Int J Mol Sci. 2024 Sep 30;25(19):10551. doi: 10.3390/ijms251910551. Int J Mol Sci. 2024. PMID: 39408877 Free PMC article. Review.
-
Structure of human TRPV4 in complex with GTPase RhoA.Nat Commun. 2023 Jun 23;14(1):3733. doi: 10.1038/s41467-023-39346-z. Nat Commun. 2023. PMID: 37353478 Free PMC article.
-
Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress.J Biol Chem. 2021 Jan-Jun;296:100171. doi: 10.1074/jbc.RA120.015059. Epub 2020 Dec 14. J Biol Chem. 2021. PMID: 33298523 Free PMC article.
-
WNK kinase is a vasoactive chloride sensor in endothelial cells.Proc Natl Acad Sci U S A. 2024 Apr 9;121(15):e2322135121. doi: 10.1073/pnas.2322135121. Epub 2024 Apr 3. Proc Natl Acad Sci U S A. 2024. PMID: 38568964 Free PMC article.
-
Platelet-derived microvesicles induce calcium oscillations and promote VSMC migration via TRPV4.Theranostics. 2021 Jan 1;11(5):2410-2423. doi: 10.7150/thno.47182. eCollection 2021. Theranostics. 2021. PMID: 33500733 Free PMC article.
References
-
- Alessandri-Haber N., Dina O. A., Joseph E. K., Reichling D., and Levine J. D. (2006) A transient receptor potential vanilloid 4–dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J. Neurosci. 26, 3864–3874 10.1523/JNEUROSCI.5385-05.2006 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
