

WDR5 Facilitates Human Cytomegalovirus Replication by Promoting Capsid Nuclear Egress
- PMID: 29437978
- PMCID: PMC5899187
- DOI: 10.1128/JVI.00207-18
WDR5 Facilitates Human Cytomegalovirus Replication by Promoting Capsid Nuclear Egress
Abstract
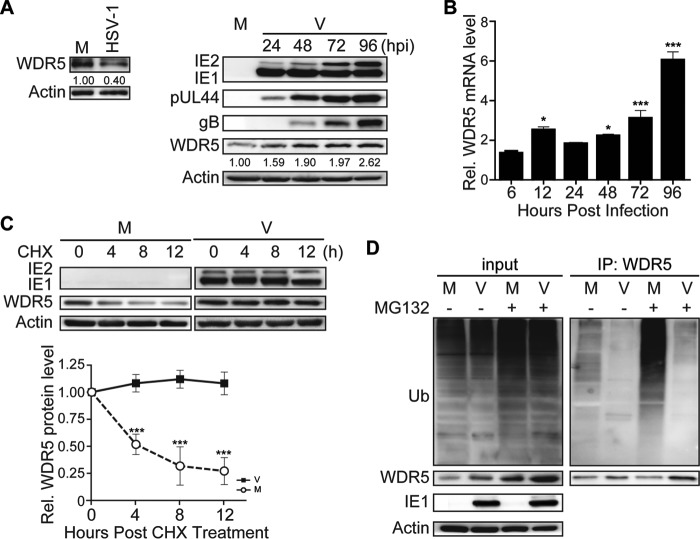
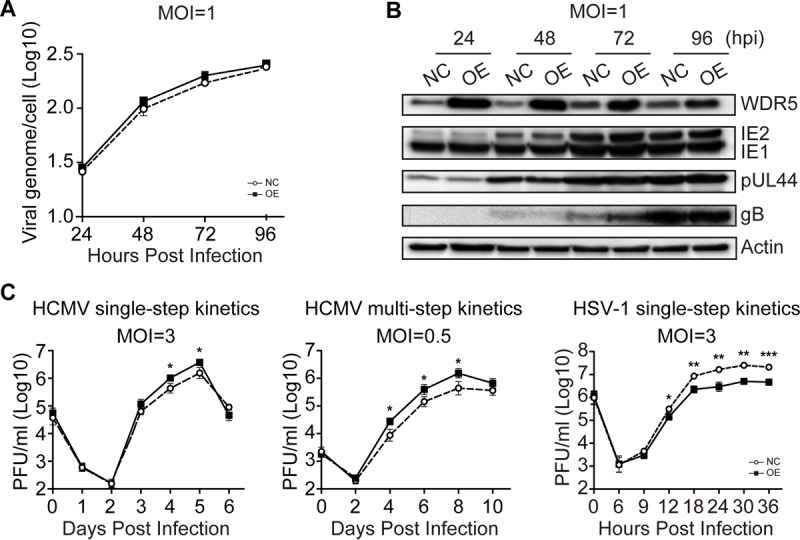
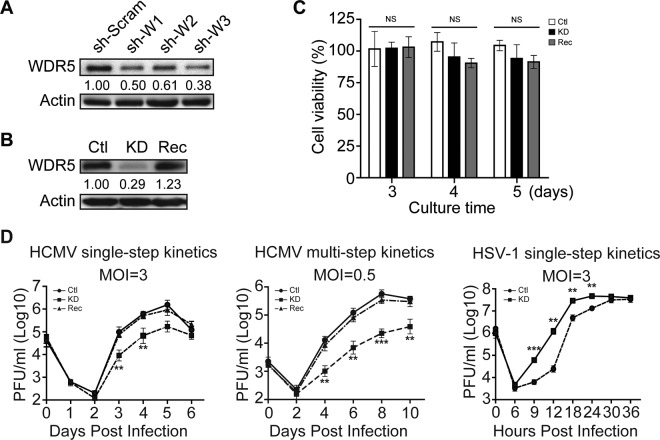
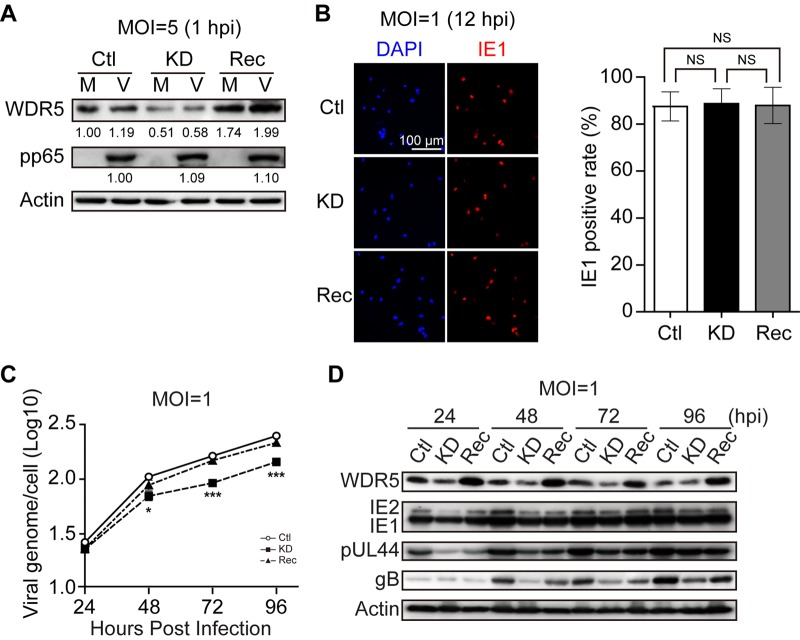
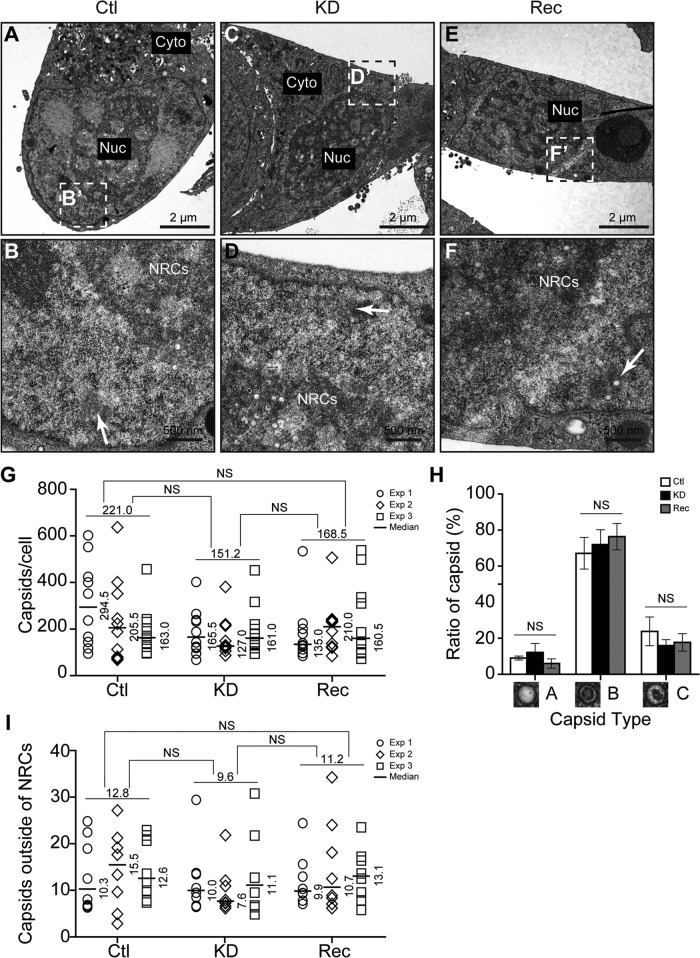
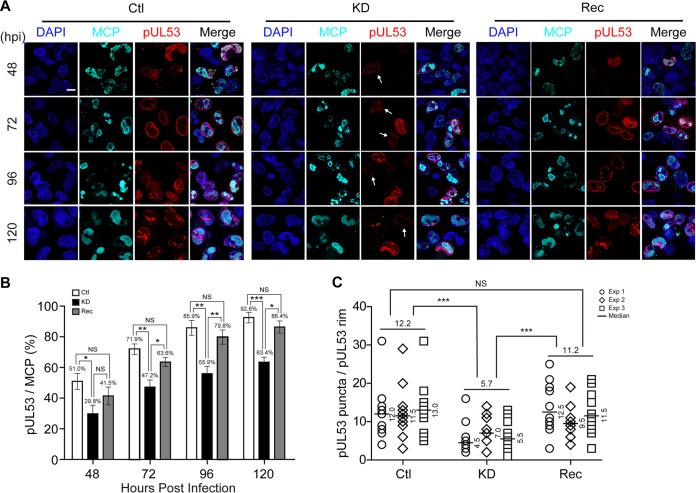
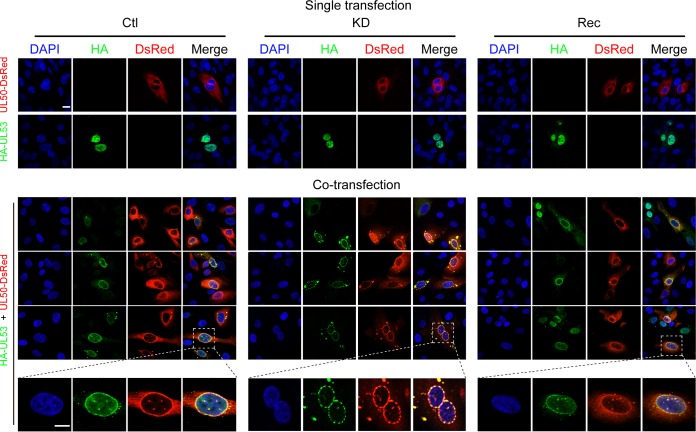
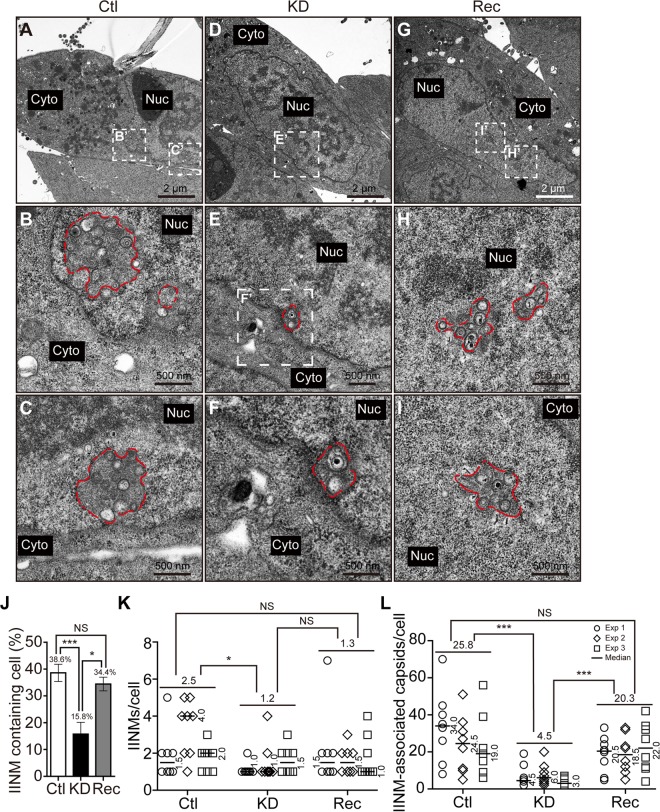
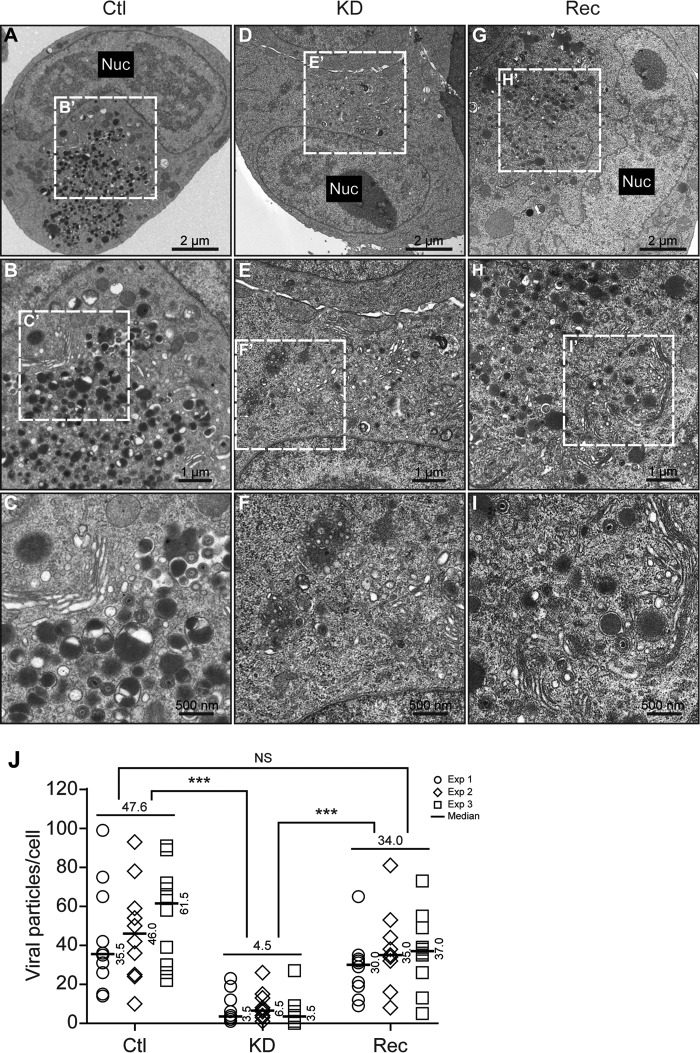
WD repeat-containing protein 5 (WDR5) is essential for assembling the VISA-associated complex to induce a type I interferon antiviral response to Sendai virus infection. However, the roles of WDR5 in DNA virus infections are not well described. Here, we report that human cytomegalovirus exploits WDR5 to facilitate capsid nuclear egress. Overexpression of WDR5 in fibroblasts slightly enhanced the infectious virus yield. However, WDR5 knockdown dramatically reduced infectious virus titers with only a small decrease in viral genome replication or gene expression. Further investigation of late steps of viral replication found that WDR5 knockdown significantly impaired formation of the viral nuclear egress complex and induced substantially fewer infoldings of the inner nuclear membrane. In addition, fewer capsids were associated with these infoldings, and there were fewer capsids in the cytoplasm. Restoration of WDR5 partially reversed these effects. These results suggest that WDR5 knockdown impairs the nuclear egress of capsids, which in turn decreases virus titers. These findings reveal an important role for a host factor whose function(s) is usurped by a viral pathogen to promote efficient replication. Thus, WDR5 represents an interesting regulatory mechanism and a potential antiviral target.IMPORTANCE Human cytomegalovirus (HCMV) has a large (∼235-kb) genome with over 170 open reading frames and exploits numerous cellular factors to facilitate its replication. HCMV infection increases protein levels of WD repeat-containing protein 5 (WDR5) during infection, overexpression of WDR5 enhances viral replication, and knockdown of WDR5 dramatically attenuates viral replication. Our results indicate that WDR5 promotes the nuclear egress of viral capsids, the depletion of WDR5 resulting in a significant decrease in production of infectious virions. This is the first report that WDR5 favors HCMV, a DNA virus, replication and highlights a novel target for antiviral therapy.
Keywords: IINMs; WDR5; capsid; human cytomegalovirus; nuclear egress.
Copyright © 2018 American Society for Microbiology.
Figures
Similar articles
-
Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions.Viruses. 2021 Dec 22;14(1):15. doi: 10.3390/v14010015. Viruses. 2021. PMID: 35062219 Free PMC article. Review.
-
A Role for Myosin Va in Human Cytomegalovirus Nuclear Egress.J Virol. 2018 Feb 26;92(6):e01849-17. doi: 10.1128/JVI.01849-17. Print 2018 Mar 15. J Virol. 2018. PMID: 29298889 Free PMC article.
-
Upon Infection, Cellular WD Repeat-Containing Protein 5 (WDR5) Localizes to Cytoplasmic Inclusion Bodies and Enhances Measles Virus Replication.J Virol. 2018 Feb 12;92(5):e01726-17. doi: 10.1128/JVI.01726-17. Print 2018 Mar 1. J Virol. 2018. PMID: 29237839 Free PMC article.
-
Localization of the WD repeat-containing protein 5 to the Virion Assembly Compartment Facilitates Human Cytomegalovirus Assembly.J Virol. 2021 Mar 25;95(8):e02101-20. doi: 10.1128/JVI.02101-20. Epub 2021 Jan 27. J Virol. 2021. PMID: 33504601 Free PMC article.
-
The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids.Rev Med Virol. 2017 Jul;27(4). doi: 10.1002/rmv.1934. Epub 2017 Jun 30. Rev Med Virol. 2017. PMID: 28664574 Review.
Cited by
-
iTRAQ-Based Proteomics Analysis of Human Cytomegalovirus Latency and Reactivation in T98G Cells.J Virol. 2022 Jan 26;96(2):e0147621. doi: 10.1128/JVI.01476-21. Epub 2021 Nov 3. J Virol. 2022. PMID: 34730396 Free PMC article.
-
Virus-host protein interactions as footprints of human cytomegalovirus replication.Curr Opin Virol. 2022 Feb;52:135-147. doi: 10.1016/j.coviro.2021.11.016. Epub 2021 Dec 16. Curr Opin Virol. 2022. PMID: 34923282 Free PMC article. Review.
-
Anterograde Viral Tracer Herpes Simplex Virus 1 Strain H129 Transports Primarily as Capsids in Cortical Neuron Axons.J Virol. 2020 Mar 31;94(8):e01957-19. doi: 10.1128/JVI.01957-19. Print 2020 Mar 31. J Virol. 2020. PMID: 31969440 Free PMC article.
-
Current Insights into the Maturation of Epstein-Barr Virus Particles.Microorganisms. 2024 Apr 17;12(4):806. doi: 10.3390/microorganisms12040806. Microorganisms. 2024. PMID: 38674750 Free PMC article. Review.
-
Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions.Viruses. 2021 Dec 22;14(1):15. doi: 10.3390/v14010015. Viruses. 2021. PMID: 35062219 Free PMC article. Review.
References
-
- Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
