

A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination
- PMID: 29401456
- PMCID: PMC5814097
- DOI: 10.1371/journal.pcbi.1005958
A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination
Abstract
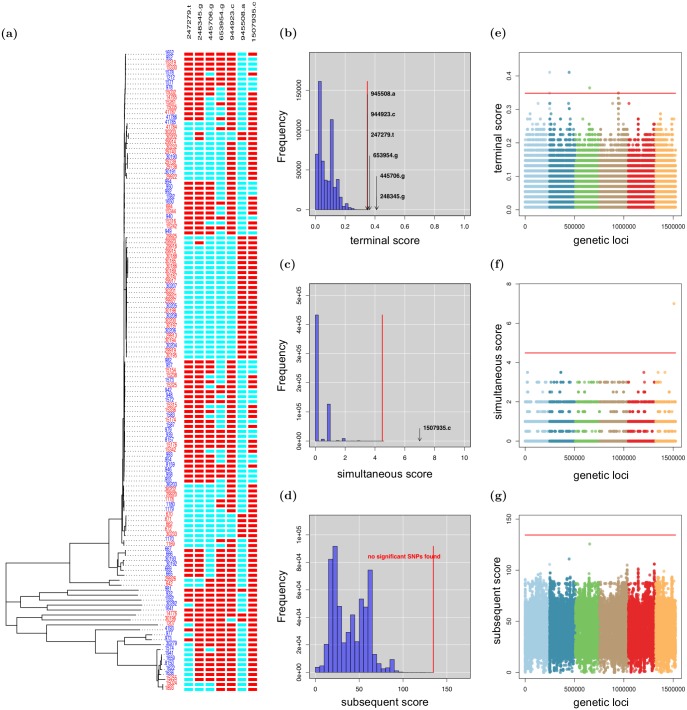
Genome-Wide Association Studies (GWAS) in microbial organisms have the potential to vastly improve the way we understand, manage, and treat infectious diseases. Yet, microbial GWAS methods established thus far remain insufficiently able to capitalise on the growing wealth of bacterial and viral genetic sequence data. Facing clonal population structure and homologous recombination, existing GWAS methods struggle to achieve both the precision necessary to reject spurious findings and the power required to detect associations in microbes. In this paper, we introduce a novel phylogenetic approach that has been tailor-made for microbial GWAS, which is applicable to organisms ranging from purely clonal to frequently recombining, and to both binary and continuous phenotypes. Our approach is robust to the confounding effects of both population structure and recombination, while maintaining high statistical power to detect associations. Thorough testing via application to simulated data provides strong support for the power and specificity of our approach and demonstrates the advantages offered over alternative cluster-based and dimension-reduction methods. Two applications to Neisseria meningitidis illustrate the versatility and potential of our method, confirming previously-identified penicillin resistance loci and resulting in the identification of both well-characterised and novel drivers of invasive disease. Our method is implemented as an open-source R package called treeWAS which is freely available at https://github.com/caitiecollins/treeWAS.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
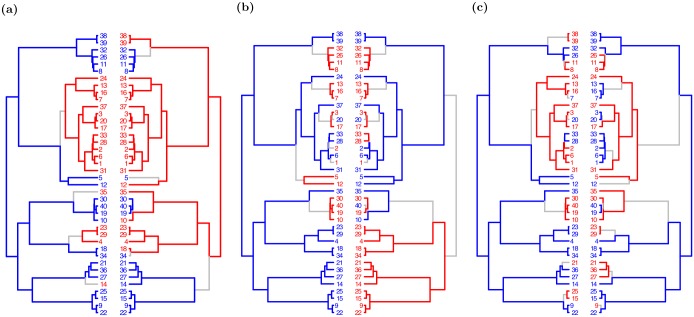
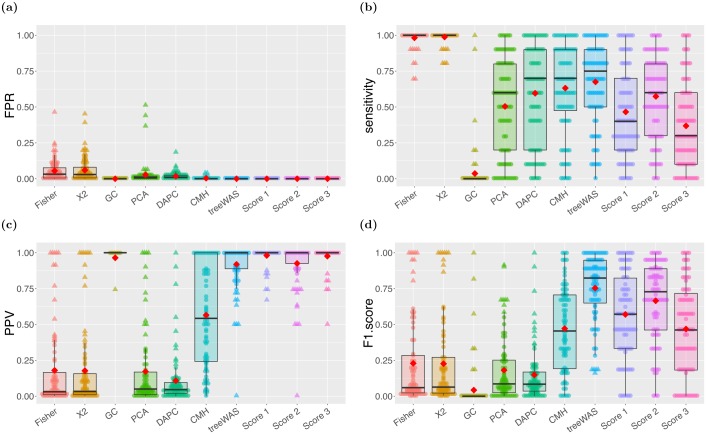
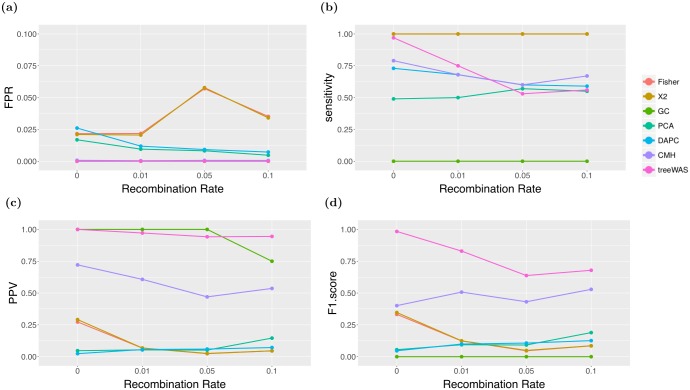
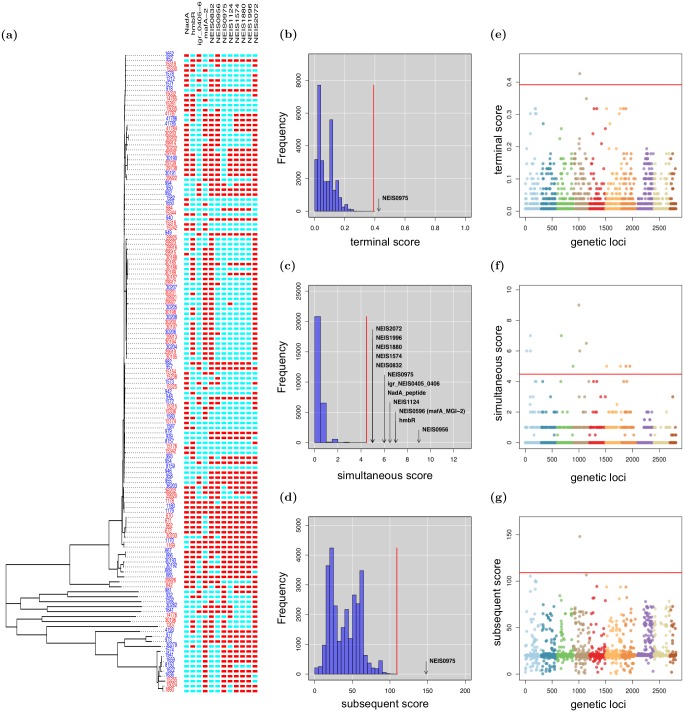
Similar articles
-
Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes.Microb Genom. 2020 Mar;6(3):e000337. doi: 10.1099/mgen.0.000337. Microb Genom. 2020. PMID: 32100713 Free PMC article.
-
RAINBOW: Haplotype-based genome-wide association study using a novel SNP-set method.PLoS Comput Biol. 2020 Feb 14;16(2):e1007663. doi: 10.1371/journal.pcbi.1007663. eCollection 2020 Feb. PLoS Comput Biol. 2020. PMID: 32059004 Free PMC article.
-
The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis.Mol Biol Evol. 1999 Jun;16(6):741-9. doi: 10.1093/oxfordjournals.molbev.a026159. Mol Biol Evol. 1999. PMID: 10368953
-
OPENMENDEL: a cooperative programming project for statistical genetics.Hum Genet. 2020 Jan;139(1):61-71. doi: 10.1007/s00439-019-02001-z. Epub 2019 Mar 26. Hum Genet. 2020. PMID: 30915546 Free PMC article. Review.
-
Population genomics: diversity and virulence in the Neisseria.Curr Opin Microbiol. 2008 Oct;11(5):467-71. doi: 10.1016/j.mib.2008.09.002. Epub 2008 Oct 14. Curr Opin Microbiol. 2008. PMID: 18822386 Free PMC article. Review.
Cited by
-
Using GWAS and Machine Learning to Identify and Predict Genetic Variants Associated with Foodborne Bacteria Phenotypic Traits.Methods Mol Biol. 2025;2852:223-253. doi: 10.1007/978-1-0716-4100-2_16. Methods Mol Biol. 2025. PMID: 39235748 Review.
-
A scalable analytical approach from bacterial genomes to epidemiology.Philos Trans R Soc Lond B Biol Sci. 2022 Oct 10;377(1861):20210246. doi: 10.1098/rstb.2021.0246. Epub 2022 Aug 22. Philos Trans R Soc Lond B Biol Sci. 2022. PMID: 35989600 Free PMC article.
-
Can machines learn the mutation signatures of SARS-CoV-2 and enable viral-genotype guided predictive prognosis?J Mol Biol. 2022 Aug 15;434(15):167684. doi: 10.1016/j.jmb.2022.167684. Epub 2022 Jun 11. J Mol Biol. 2022. PMID: 35700770 Free PMC article.
-
Genomic correlates of extraintestinal infection are linked with changes in cell morphology in Campylobacter jejuni.Microb Genom. 2019 Feb;5(2):e000251. doi: 10.1099/mgen.0.000251. Epub 2019 Feb 19. Microb Genom. 2019. PMID: 30777818 Free PMC article.
-
Comparative genomics in infectious disease.Curr Opin Microbiol. 2020 Feb;53:61-70. doi: 10.1016/j.mib.2020.02.009. Epub 2020 Apr 2. Curr Opin Microbiol. 2020. PMID: 32248056 Free PMC article. Review.
References
-
- WHO. World Health Statistics Global Health Indicators: Cause-specific mortality and morbidity. World Health Organisation; 2015;p. 72.
-
- Lowder BV, Guinane CM, Ben Zakour NL, Weinert LA, Conway-Morris A, Cartwright RA, et al. Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2009. 17 November;106(46):19545–19550. doi: 10.1073/pnas.0909285106 - DOI - PMC - PubMed
-
- Guinane CM, Ben Zakour NL, Tormo-Mas MA, Weinert LA, Lowder BV, Cartwright RA, et al. Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol Evol. 2010. 12 July;2:454–466. doi: 10.1093/gbe/evq031 - DOI - PMC - PubMed
-
- Kiechle FL, Zhang X, Holland-Staley CA. The -omics era and its impact. Arch Pathol Lab Med. 2004. December;128(12):1337–1345. - PubMed
-
- Holden MTG, Hsu LY, Kurt K, Weinert LA, Mather AE, Harris SR, et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 2013. April;23(4):653–664. doi: 10.1101/gr.147710.112 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
