

Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks
- PMID: 29392184
- PMCID: PMC5785775
- DOI: 10.1021/acscentsci.7b00512
Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks
Abstract
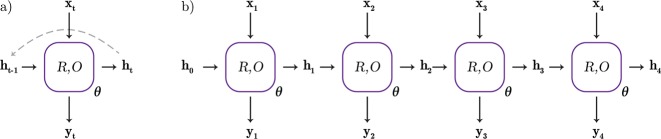
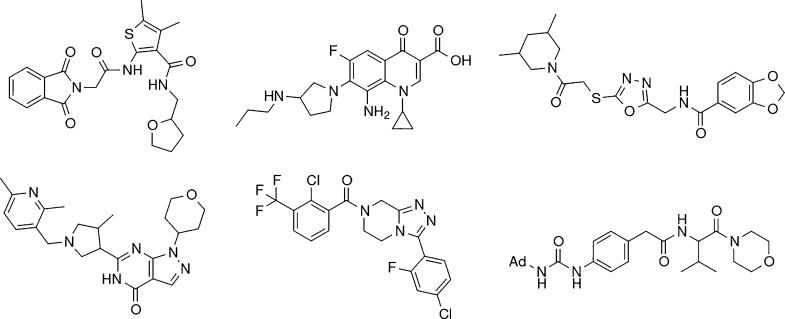
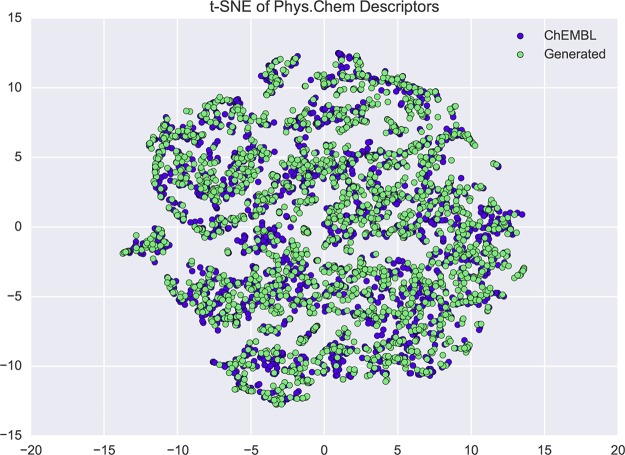
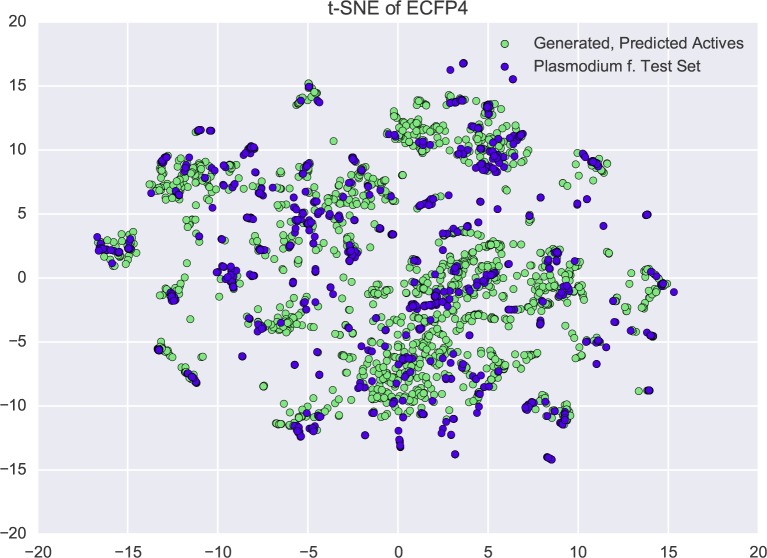
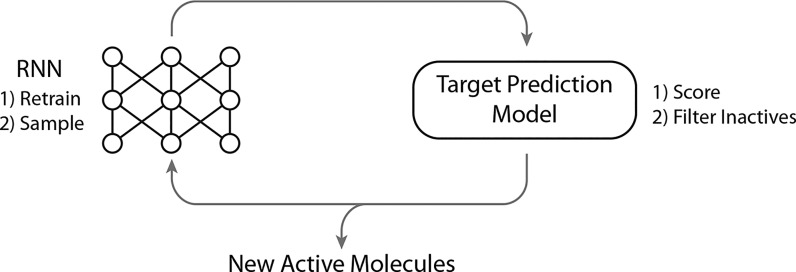
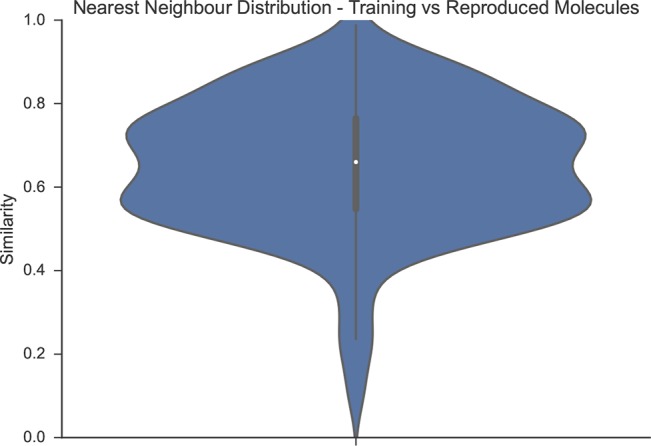
In de novo drug design, computational strategies are used to generate novel molecules with good affinity to the desired biological target. In this work, we show that recurrent neural networks can be trained as generative models for molecular structures, similar to statistical language models in natural language processing. We demonstrate that the properties of the generated molecules correlate very well with the properties of the molecules used to train the model. In order to enrich libraries with molecules active toward a given biological target, we propose to fine-tune the model with small sets of molecules, which are known to be active against that target. Against Staphylococcus aureus, the model reproduced 14% of 6051 hold-out test molecules that medicinal chemists designed, whereas against Plasmodium falciparum (Malaria), it reproduced 28% of 1240 test molecules. When coupled with a scoring function, our model can perform the complete de novo drug design cycle to generate large sets of novel molecules for drug discovery.
Conflict of interest statement
The authors declare no competing financial interest.
Figures






Similar articles
-
Generation of focused drug molecule library using recurrent neural network.J Mol Model. 2023 Nov 6;29(12):361. doi: 10.1007/s00894-023-05772-5. J Mol Model. 2023. PMID: 37932607
-
Molecular de-novo design through deep reinforcement learning.J Cheminform. 2017 Sep 4;9(1):48. doi: 10.1186/s13321-017-0235-x. J Cheminform. 2017. PMID: 29086083 Free PMC article.
-
Deep reinforcement learning for de novo drug design.Sci Adv. 2018 Jul 25;4(7):eaap7885. doi: 10.1126/sciadv.aap7885. eCollection 2018 Jul. Sci Adv. 2018. PMID: 30050984 Free PMC article.
-
Training recurrent neural networks as generative neural networks for molecular structures: how does it impact drug discovery?Expert Opin Drug Discov. 2022 Oct;17(10):1071-1079. doi: 10.1080/17460441.2023.2134340. Epub 2022 Oct 17. Expert Opin Drug Discov. 2022. PMID: 36216812 Review.
-
Generative Deep Learning for Targeted Compound Design.J Chem Inf Model. 2021 Nov 22;61(11):5343-5361. doi: 10.1021/acs.jcim.0c01496. Epub 2021 Oct 26. J Chem Inf Model. 2021. PMID: 34699719 Review.
Cited by
-
Generative deep learning enables the discovery of a potent and selective RIPK1 inhibitor.Nat Commun. 2022 Nov 12;13(1):6891. doi: 10.1038/s41467-022-34692-w. Nat Commun. 2022. PMID: 36371441 Free PMC article.
-
A Generative Neural Network for Maximizing Fitness and Diversity of Synthetic DNA and Protein Sequences.Cell Syst. 2020 Jul 22;11(1):49-62.e16. doi: 10.1016/j.cels.2020.05.007. Epub 2020 Jun 25. Cell Syst. 2020. PMID: 32711843 Free PMC article.
-
Active Learning and the Potential of Neural Networks Accelerate Molecular Screening for the Design of a New Molecule Effective against SARS-CoV-2.Biomed Res Int. 2021 May 25;2021:6696012. doi: 10.1155/2021/6696012. eCollection 2021. Biomed Res Int. 2021. PMID: 34124259 Free PMC article.
-
Targeting ion channels with ultra-large library screening for hit discovery.Front Mol Neurosci. 2024 Jan 5;16:1336004. doi: 10.3389/fnmol.2023.1336004. eCollection 2023. Front Mol Neurosci. 2024. PMID: 38249296 Free PMC article. Review.
-
Geometry-Complete Diffusion for 3D Molecule Generation and Optimization.ArXiv [Preprint]. 2024 May 24:arXiv:2302.04313v6. ArXiv. 2024. Update in: Commun Chem. 2024 Jul 3;7(1):150. doi: 10.1038/s42004-024-01233-z PMID: 36798459 Free PMC article. Updated. Preprint.
References
-
- Reymond J.-L.; Ruddigkeit L.; Blum L.; van Deursen R. The enumeration of chemical space. Wiley Interdisc. Rev. Comp. Mol. Sci. 2012, 2, 717–733. 10.1002/wcms.1104. - DOI
-
- Schneider G.; Baringhaus K.-H.. Molecular design: concepts and applications; John Wiley & Sons: 2008.
-
- Stumpfe D.; Bajorath J. Similarity searching. Wiley Interdisc. Rev. Comp. Mol. Sci. 2011, 1, 260–282. 10.1002/wcms.23. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
