

Metatranscriptome of human faecal microbial communities in a cohort of adult men
- PMID: 29335555
- PMCID: PMC6557121
- DOI: 10.1038/s41564-017-0084-4
Metatranscriptome of human faecal microbial communities in a cohort of adult men
Abstract
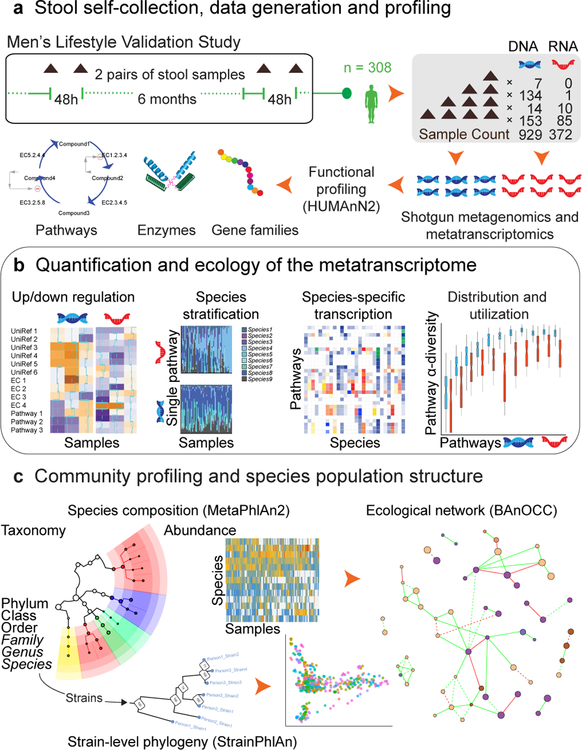
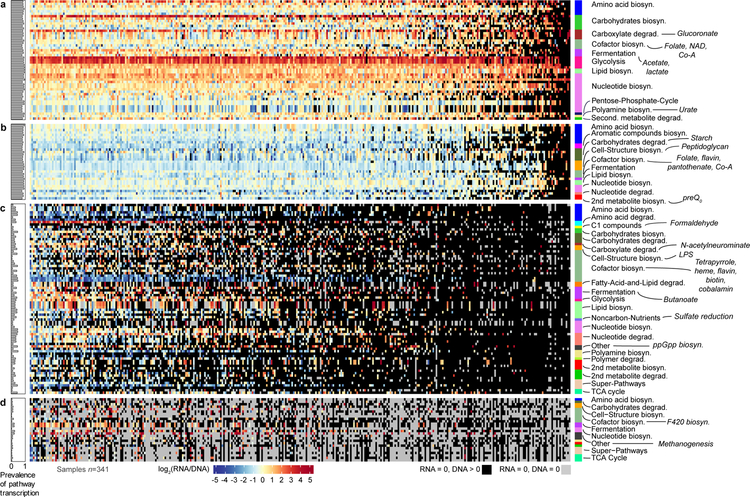
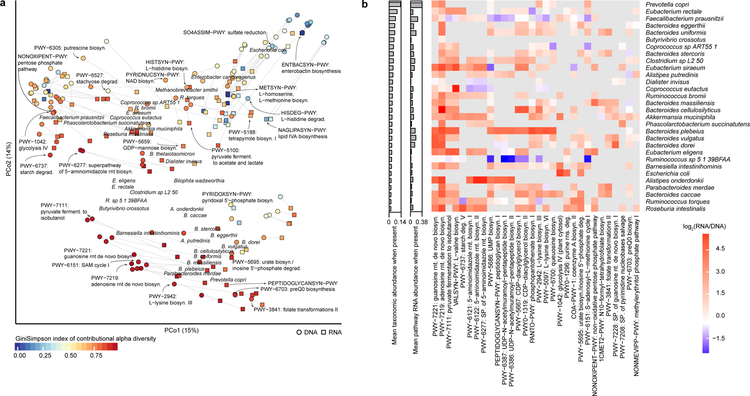
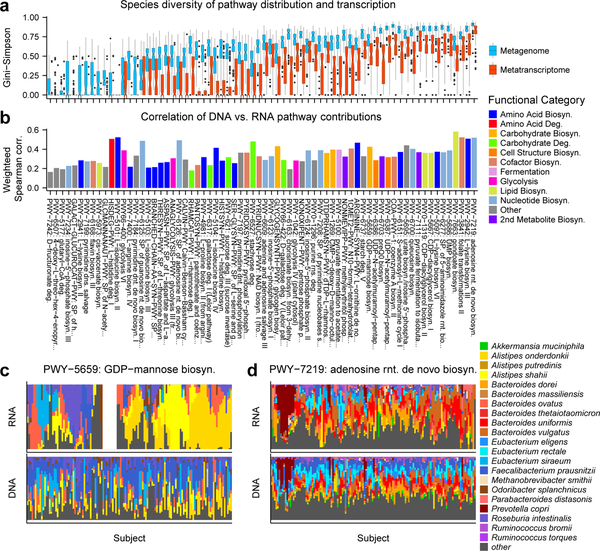
The gut microbiome is intimately related to human health, but it is not yet known which functional activities are driven by specific microorganisms' ecological configurations or transcription. We report a large-scale investigation of 372 human faecal metatranscriptomes and 929 metagenomes from a subset of 308 men in the Health Professionals Follow-Up Study. We identified a metatranscriptomic 'core' universally transcribed over time and across participants, often by different microorganisms. In contrast to the housekeeping functions enriched in this core, a 'variable' metatranscriptome included specialized pathways that were differentially expressed both across participants and among microorganisms. Finally, longitudinal metagenomic profiles allowed ecological interaction network reconstruction, which remained stable over the six-month timespan, as did strain tracking within and between participants. These results provide an initial characterization of human faecal microbial ecology into core, subject-specific, microorganism-specific and temporally variable transcription, and they differentiate metagenomically versus metatranscriptomically informative aspects of the human faecal microbiome.
Conflict of interest statement
Competing interests
The authors declare no competing financial interests.
Figures
Similar articles
-
Stability of the human faecal microbiome in a cohort of adult men.Nat Microbiol. 2018 Mar;3(3):347-355. doi: 10.1038/s41564-017-0096-0. Epub 2018 Jan 15. Nat Microbiol. 2018. PMID: 29335554 Free PMC article.
-
Functional dynamics of bacterial species in the mouse gut microbiome revealed by metagenomic and metatranscriptomic analyses.PLoS One. 2020 Jan 24;15(1):e0227886. doi: 10.1371/journal.pone.0227886. eCollection 2020. PLoS One. 2020. PMID: 31978162 Free PMC article.
-
Relating the metatranscriptome and metagenome of the human gut.Proc Natl Acad Sci U S A. 2014 Jun 3;111(22):E2329-38. doi: 10.1073/pnas.1319284111. Epub 2014 May 19. Proc Natl Acad Sci U S A. 2014. PMID: 24843156 Free PMC article.
-
Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes.Brief Bioinform. 2018 Nov 27;19(6):1415-1429. doi: 10.1093/bib/bbx051. Brief Bioinform. 2018. PMID: 28481971 Review.
-
The human gut microbiome, a taxonomic conundrum.Syst Appl Microbiol. 2015 Jun;38(4):276-86. doi: 10.1016/j.syapm.2015.03.004. Epub 2015 Mar 25. Syst Appl Microbiol. 2015. PMID: 25864640 Review.
Cited by
-
A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases.Proc Natl Acad Sci U S A. 2021 Jun 8;118(23):e2023202118. doi: 10.1073/pnas.2023202118. Epub 2021 Jun 3. Proc Natl Acad Sci U S A. 2021. PMID: 34083435 Free PMC article.
-
Engraftment of essential functions through multiple fecal microbiota transplants in chronic antibiotic-resistant pouchitis-a case study using metatranscriptomics.Microbiome. 2023 Dec 1;11(1):269. doi: 10.1186/s40168-023-01713-9. Microbiome. 2023. PMID: 38037086 Free PMC article.
-
Human gut bacteria contain acquired interbacterial defence systems.Nature. 2019 Nov;575(7781):224-228. doi: 10.1038/s41586-019-1708-z. Epub 2019 Oct 30. Nature. 2019. PMID: 31666699 Free PMC article.
-
Intraintestinal Analysis of the Functional Activity of Microbiomes and Its Application to the Common Marmoset Intestine.mSystems. 2022 Oct 26;7(5):e0052022. doi: 10.1128/msystems.00520-22. Epub 2022 Aug 25. mSystems. 2022. PMID: 36005400 Free PMC article.
-
Nutritional redundancy in the human diet and its application in phenotype association studies.Nat Commun. 2023 Jul 18;14(1):4316. doi: 10.1038/s41467-023-39836-0. Nat Commun. 2023. PMID: 37463879 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
