

So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again!
- PMID: 29328962
- PMCID: PMC6037615
- DOI: 10.1016/j.sbi.2017.12.004
So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again!
Abstract
Glycosaminoglycans (GAGs) play key roles in virtually all biologic responses through their interaction with proteins. A major challenge in understanding these roles is their massive structural complexity. Computational approaches are extremely useful in navigating this bottleneck and, in some cases, the only avenue to gain comprehensive insight. We discuss the state-of-the-art on computational approaches and present a flowchart to help answer most basic, and some advanced, questions on GAG-protein interactions. For example, firstly, does my protein bind to GAGs?; secondly, where does the GAG bind?; thirdly, does my protein preferentially recognize a particular GAG type?; fourthly, what is the most optimal GAG chain length?; fifthly, what is the structure of the most favored GAG sequence?; and finally, is my GAG-protein system 'specific', 'non-specific', or a combination of both? Recent advances show the field is now poised to enable a non-computational researcher perform advanced experiments through the availability of various tools and online servers.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Conflict of interest statement
The authors have no conflicts of interest to declare
Figures
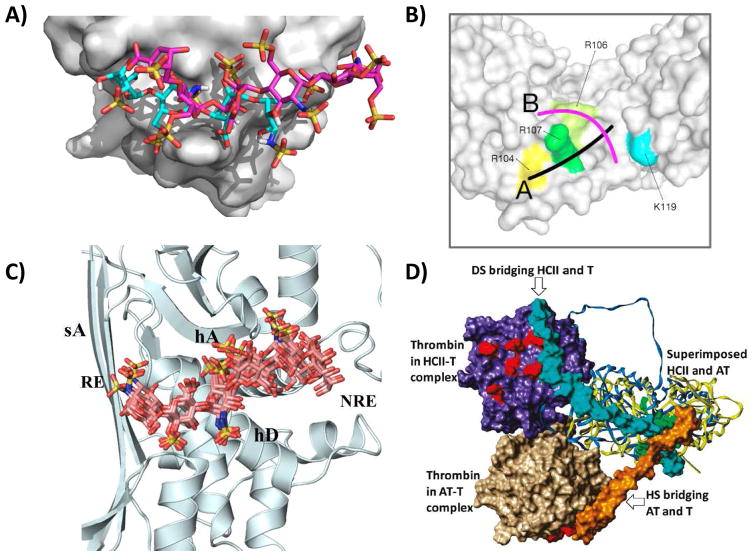
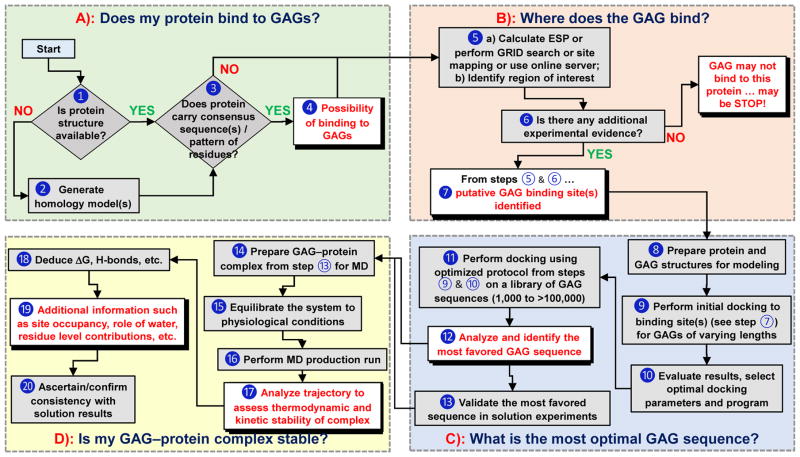
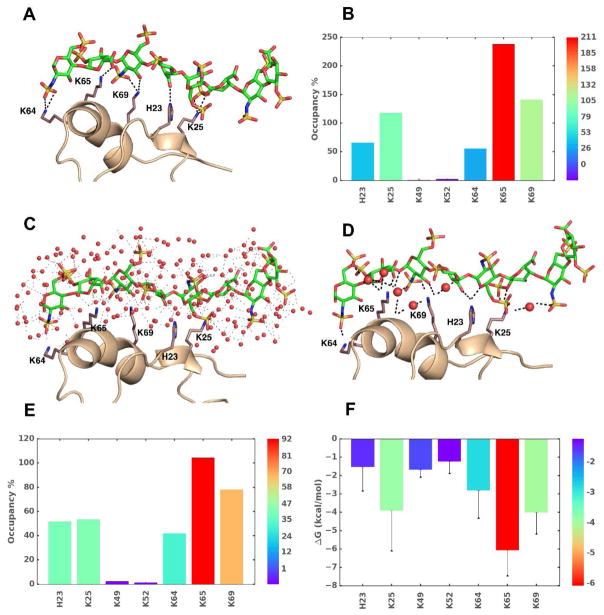
Similar articles
-
Computational Analysis of Solvent Inclusion in Docking Studies of Protein-Glycosaminoglycan Systems.Methods Mol Biol. 2018;1762:445-454. doi: 10.1007/978-1-4939-7756-7_22. Methods Mol Biol. 2018. PMID: 29594785
-
Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins.Bioessays. 1998 Feb;20(2):156-67. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. Bioessays. 1998. PMID: 9631661 Review.
-
Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy.Molecules. 2018 Sep 11;23(9):2314. doi: 10.3390/molecules23092314. Molecules. 2018. PMID: 30208595 Free PMC article. Review.
-
The structure of glycosaminoglycans and their interactions with proteins.Chem Biol Drug Des. 2008 Dec;72(6):455-82. doi: 10.1111/j.1747-0285.2008.00741.x. Chem Biol Drug Des. 2008. PMID: 19090915 Review.
-
Specificity of glycosaminoglycan-protein interactions.Curr Opin Struct Biol. 2018 Jun;50:101-108. doi: 10.1016/j.sbi.2017.12.011. Epub 2018 Feb 9. Curr Opin Struct Biol. 2018. PMID: 29455055 Review.
Cited by
-
Discovery of Sulfated Small Molecule Inhibitors of Matrix Metalloproteinase-8.Biomolecules. 2020 Aug 9;10(8):1166. doi: 10.3390/biom10081166. Biomolecules. 2020. PMID: 32784891 Free PMC article.
-
Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease.Front Mol Biosci. 2021 Mar 9;8:639666. doi: 10.3389/fmolb.2021.639666. eCollection 2021. Front Mol Biosci. 2021. PMID: 33768117 Free PMC article. Review.
-
Role of Glycosaminoglycans in Procathepsin B Maturation: Molecular Mechanism Elucidated by a Computational Study.J Chem Inf Model. 2020 Apr 27;60(4):2247-2256. doi: 10.1021/acs.jcim.0c00023. Epub 2020 Apr 1. J Chem Inf Model. 2020. PMID: 32155059 Free PMC article.
-
Computational toolbox for the analysis of protein-glycan interactions.Beilstein J Org Chem. 2024 Aug 22;20:2084-2107. doi: 10.3762/bjoc.20.180. eCollection 2024. Beilstein J Org Chem. 2024. PMID: 39189002 Free PMC article. Review.
-
GAG Builder: a web-tool for modeling 3D structures of glycosaminoglycans.Glycobiology. 2019 Jul 1;29(7):515-518. doi: 10.1093/glycob/cwz027. Glycobiology. 2019. PMID: 31034567 Free PMC article.
References
-
- Balagurunathan K, Nakato H, Desai UR. Glycosaminoglycans. Chemistry and Biology. Methods Mol Biol. 2015;1229:1–625. - PubMed
-
- Mulloy B, Hogwood J, Gray E, Lever R, Page CP. Pharmacology of Heparin and Related Drugs. Pharmacol Rev. 2016;68:76–141. - PubMed
-
- Joseph PR, Mosier PD, Desai UR, Rajarathnam K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem J. 2015;472:121–133. The paper presents a good combination of biophysical and computational experiments to understand the nature of interactions in monomer–dimer equilibrium. MD studies show that the GAG binding is plastic and pinpointed residues contributing to the dynamic nature of the process. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
